


Rewriting hepatic fate: emerging gene therapy strategies for liver disease
 0
0 
Abstract
Several anatomical and physiological features make the liver particularly suitable for gene-based therapeutic strategies, including its extensive vascularization, fenestrated sinusoidal endothelium, and high metabolic capacity. In the past decade, liver-directed gene therapies have evolved from experimental concepts to clinical applications for various inherited and systemic disorders. Adeno-associated viral vectors, lentiviral systems, and lipid nanoparticles are currently the main platforms for delivering therapeutic genes and genome-editing tools to hepatocytes. Genome editing technologies such as CRISPR-Cas nucleases, base editors, and prime editors have enabled more precise modification of endogenous loci. Early clinical studies in disorders like hemophilia, transthyretin amyloidosis, ornithine transcarbamylase deficiency, and Crigler-Najjar syndrome show that partial correction of hepatic gene function can lead to meaningful clinical benefits. However, several challenges hinder broader clinical implementation, including immune responses to viral capsids and nanoparticle components, dose-dependent toxicity, limited packaging capacity of some vectors, and uncertainties about long-term safety and durability, especially in pediatric populations. Efficient and cell-type-specific delivery beyond hepatocytes remains a major challenge, particularly for diseases driven by non-parenchymal cells or malignant transformation. This article discusses recent advancements in delivery technologies and genome editing approaches for liver disease, as well as current translational barriers and emerging strategies aimed at enhancing specificity, durability, and safety. Collectively, these advances suggest that increasingly precise and programmable gene-based interventions may play a central role in future therapies for a broad spectrum of liver diseases.
Keywords
INTRODUCTION
The liver occupies a unique position at the intersection of metabolism, immunity, and systemic homeostasis[1]. It plays a crucial role in synthesizing most circulating proteins, regulating glucose and lipid levels, processing nutrients and toxins from the gut, and detoxifying harmful substances. There are various genetic liver disorders [Table 1], such as urea cycle disorders, Crigler-Najjar syndrome, Wilson disease, and different glycogen storage diseases that often present in early childhood with severe symptoms[2]. Acquired liver conditions associated with genetic alterations typically develop over years and can result in cirrhosis, portal hypertension, and hepatocellular carcinoma (HCC). Although orthotopic liver transplantation is the primary treatment for many advanced liver diseases, it is hindered by organ shortages, surgical risks, and the need for lifelong immunosuppression.
Inherited genetic liver diseases-causative gene (OMIM accession) and principal hepatic phenotype
| Disease | Causative gene | Location | OMIM1 | Liver-related phenotype |
| Wilson disease | ATP7B | 13q14.3 | 606882 | Progressive copper accumulation leads to hepatic steatosis, fibrosis and cirrhosis, often accompanied by neuro-psychiatric signs and Kayser-Fleischer rings |
| Hereditary hemochromatosis (type 1) | HFE | 6p22.2 | 613609 | Iron overload causes hepatomegaly, hepatic fibrosis, cirrhosis, and an increased risk of hepatocellular carcinoma (HCC) |
| Hemochromatosis (type 2A); hemojuvelin deficiency | HJV | 1q21.1 | 608374 | Juvenile iron overload causes aggressive liver injury in childhood |
| Hemochromatosis (type 2B); hepcidin deficiency | HAMP | 19q13.12 | 606464 | Early-onset severe iron overload results in rapid progression to cirrhosis |
| Hemochromatosis (type 3); transferrin receptor2 disease | TFR2 | 7q22.1 | 604720 | Moderate iron overload leads to hepatic fibrosis, which is milder than HFE disease |
| Hemochromatosis (type 4); ferroportin disease | SLC40A1 | 2q32.2 | 604653 | Autosomal dominant iron loading results in variable hepatic fibrosis and sometimes anemia of chronic disease |
| Progressive familial intrahepatic cholestasis 1 (PFIC1), previously known as Byler disease | ATP8B1 | 18q21.31 | 211600 | Low-GGT cholestasis, neonatal jaundice, progressive fibrosis, and possible liver failure |
| PFIC2 | ABCB11 | 2q31.1 | 601847 | Severe cholestasis present with low GGT, high transaminases, and early liver failure |
| PFIC3 | ABCB4 | 7q21.12 | 602347 | High GGT cholestasis caused by ABCB4 deficiency leading to biliary fibrosis, gallstones, and cholangiopathy |
| PFIC4 | TJP2 | 9q21.11 | 607709 | Neonatal cholestasis results in progressive fibrosis and occasional extra-hepatic signs |
| DubinJohnson syndrome | ABCC2 | 10q24.2 | 601107 | Conjugated hyperbilirubinemia with black liver pigment is clinically benign |
| Rotor syndrome | SLCO1B1/ SLCO1B3 | 12p12.1/12p12.2 | 604843/605495 | Conjugated hyperbilirubinemia is due to impaired hepatic uptake without liver injury |
| Alagille syndrome | JAG1/NOTCH2 | 20p12.2/1p12 | 601920/600275 | Bile-duct paucity leads to cholestasis, cardiac anomalies, and characteristic facial/vertebral features |
| Low-phospholipid-associated cholelithiasis (GBD1/LPAC) | ABCB4 | 7q21.12 | 600803 | Cholesterol gallstones, intra-hepatic cholestasis of pregnancy, and elevated liver enzymes are common |
| Crigler-Najjar syndrome (type I) or Gilbert-type unconjugated hyperbilirubinemia and (type II) | UGT1A1 | 2q37.1 | 191740 | Deficiency of bilirubin-UDP-glucuronosyltransferase presents in severe (type I) or moderate (type II) forms. Type I manifests in the neonatal period with jaundice and a high risk of kernicterus, requiring phototherapy and liver transplantation to cure. Type II shows lower bilirubin levels and typically responds to phenobarbital |
| Cystic fibrosis (hepatic phenotype) | CFTR | 7q31.2 | 602421 | Biliary fibrosis, focal biliary cirrhosis, and fat malabsorption result in liver disease in approximately 30% of patients |
Against this backdrop, the potential to correct the molecular root causes of liver disease through gene therapy has been compelling for decades. The liver is anatomically and physiologically accessible to systemically administered vectors[3,4], and even partial restoration of function in a subset of hepatocytes can lead to significant clinical benefits. Early gene therapy efforts focused on liver-directed gene addition for systemic diseases such as hemophilia, where hepatocytes act as a biofactory for coagulation factors[4,5]. These pioneering trials established key principles regarding dosing, vector selection, and immune responses, while also highlighting the limitations of early approaches. Advances in human genetics and functional genomics have expanded the catalog of causal variants and disease-modifying loci associated with liver disease, emphasizing the diverse roles of hepatocytes, cholangiocytes, immune cells, and stromal populations in disease pathogenesis[3,4].
Recent progress in liver gene therapy has been driven by three major developments. Firstly, vector technology has advanced substantially, particularly with hepatotropic adeno-associated viral (AAV) serotypes and the growing clinical use of non-viral lipid nanoparticle (LNP) delivery systems as a proven method for delivering nucleic acids to the liver[6-8]. Secondly, genome editing technologies, such as clustered regularly interspaced short palindromic repeats (CRISPR)-associated (Cas) nucleases, base editors, and prime editors, now allow for precise modifications of endogenous loci with high efficiency in the liver[9]. Thirdly, high-throughput genomics, single-cell profiling, and spatial transcriptomics are providing detailed insights into the molecular landscape of both healthy and diseased liver tissue, revealing new therapeutic targets and identifying the specific cell types that need to be targeted in different disease contexts. Together, these developments are shifting the field away from a “one transgene-one disease” paradigm toward modular and customizable interventions that can be adapted to different conditions and tailored to individual patients[7].
This article first examines the biological features that make the liver an attractive yet challenging target for gene therapy. It then reviews the major delivery platforms for liver-directed interventions and discusses their translational advantages and limitations. Subsequent sections focus on therapeutic strategies, moving from traditional gene addition toward precision genome editing and expanding the discussion beyond hepatocytes to non-parenchymal cell types involved in fibrosis, inflammation, and cholestasis. Lessons from early clinical programs in monogenic metabolic and cholestatic liver diseases, as well as liver cancer, are then highlighted. The subsequent section outlines key challenges and safety concerns, including immunogenicity, genotoxicity, re-dosing, pediatric applications, and manufacturing and access issues. Finally, future directions will be discussed, including next-generation vectors, safer and more versatile editing modalities, regenerative and reprogramming approaches, combination therapies, as well as ethical and societal considerations. The ultimate goal is to provide a forward-looking perspective on how gene therapy could be integrated into the evolving therapeutic landscape of hepatology and how emerging technologies might allow us to fundamentally change the natural course of liver disease. At the same time, it is important to critically assess both the opportunities and current limitations of these approaches, as many technologies are still in the early stages of clinical translation.
The LIVER AS A TARGET FOR GENE THERAPY
Several biological and anatomical features make the liver an attractive target for gene-based interventions, including the lobular microarchitecture and the division of labor between hepatocytes and non-parenchymal cells illustrated in Figure 1. The liver receives substantial blood flow from the hepatic artery and portal vein, which supply oxygenated blood and nutrient-rich venous return from the intestine. The hepatic microvasculature consists of low-pressure sinusoids lined by specialized liver sinusoidal endothelial cells (LSECs) that are fenestrated and lack a continuous basement membrane[10]. These fenestrations, combined with low shear stress and a large sinusoidal surface area, allow for efficient exchange of macromolecules between blood and hepatocytes[11,12]. Consequently, systemically administered viral vectors and nanoparticles can access hepatocytes with high efficiency compared to many other tissues.
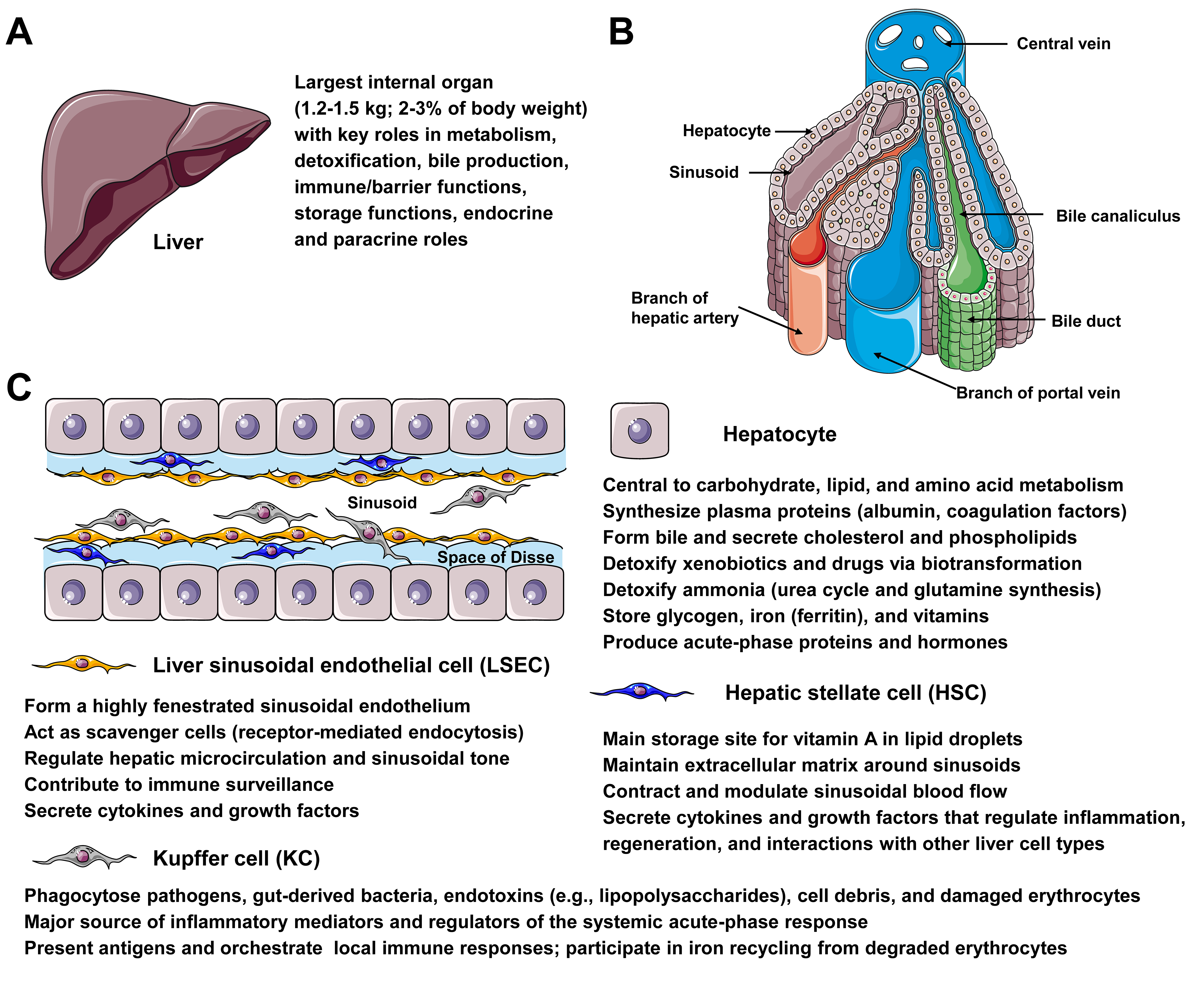
Figure 1. Microanatomy and major liver cell types relevant for gene therapy. (A) The liver, weighing 1.2-1.5 kg and accounting for 2%-3% of body weight, plays vital roles in metabolism, detoxification, bile production, immune/barrier functions, storage, and endocrine/paracrine signaling, making it an attractive target for gene therapy. (B) The diagram shows a hepatic lobule, emphasizing the intralobular (central) vein, sinusoids, bile canaliculi, bile ducts, branches of the portal vein and hepatic artery, and the positioning of hepatocytes. (C) The diagram depicts the architecture of the liver sinusoid, which is lined by fenestrated endothelia and interspersed with Kupffer cells (KCs). Hepatic stellate cells (HSC) are located within the narrow space of Disse, formed by a layer of liver sinusoidal cells (LSECs) and cords of hepatocytes. Hepatocytes play key physiological functions, including central roles in carbohydrate, lipid, and amino acid metabolism; synthesis of plasma proteins (e.g., albumin and coagulation factors); formation of bile and secretion of cholesterol and phospholipids; detoxification of xenobiotics and ammonia; storage of glycogen, iron, and vitamins; and production of acute-phase proteins and hormones. LSECs form a highly fenestrated sinusoidal endothelium, act as scavenger cells via receptor-mediated endocytosis, regulate hepatic microcirculation and sinusoidal tone, and contribute to immune surveillance. HSCs are the main storage site for vitamin A in lipid droplets, maintain the extracellular matrix around sinusoids, contract to modulate sinusoidal blood flow, and secrete cytokines and growth factors that regulate inflammation and regeneration. KCs phagocytose pathogens, gut-derived bacteria, endotoxins, cell debris, and damaged erythrocytes, produce inflammatory mediators that shape the acute-phase response, present antigens, orchestrate local immune responses, and participate in iron recycling.
In addition to accessibility, the functional redundancy and regenerative capacity of the liver provide therapeutic advantages[13,14]. Because of this threshold effect, therapeutic benefit may occur even when only a fraction of hepatocytes are genetically corrected. Furthermore, hepatocytes can proliferate after injury or partial hepatectomy, allowing corrected cells to expand and potentially repopulate large areas of the liver parenchyma under the control of autocrine and paracrine signals[13-15]. This phenomenon has been demonstrated in preclinical models in which gene-corrected hepatocytes or hepatocyte-like cells are transplanted and selectively expand when they confer a survival advantage.
However, the same features that make the liver an appealing target also introduce specific challenges. The liver plays a central role in both innate and adaptive immunity, acting as a filter for gut-derived antigens and hosting resident immune cells such as Kupffer cells, innate lymphoid cells, and various T cell subsets[16]. Viral vectors or nanoparticles entering the hepatic circulation encounter these cells, which can mediate rapid clearance, complement activation, and cytokine release[17]. Moreover, hepatocytes themselves can present antigenic peptides derived from vector capsids or transgene products, potentially eliciting cytotoxic T cell responses that attenuate or eliminate transduced cells[18]. This immunological environment must therefore be carefully considered when designing liver-directed gene therapies.
Disease stage also influences the suitability of the liver as a therapeutic target. In early disease, the vascular architecture and hepatocyte function may be relatively preserved, favoring efficient vector uptake and robust transgene expression[19]. In contrast, advanced fibrosis and cirrhosis are characterized by architectural distortion, capillarization of sinusoids, increased extracellular matrix deposition, and changes in cell-type abundance and phenotype[20]. These changes can reduce vector access to hepatocytes, alter biodistribution between parenchymal and non-parenchymal cells, and influence cellular responses to transduction and editing. In progressive diseases, the timing of intervention relative to the onset of irreversible damage becomes a critical consideration[19].
Another important dimension is the diversity of liver-resident cell types and their roles in disease. While hepatocytes are the main parenchymal cells and the principal site of many metabolic functions, non-parenchymal populations play key roles in pathogenesis[20,21]. Cholangiocytes line the biliary tree and are central in cholestatic diseases such as primary sclerosing cholangitis and biliary atresia. Hepatic stellate cells (HSCs) are the main collagen-producing cells in fibrogenesis, and their activation drives scar formation[20,21]. LSECs regulate portal pressure, leukocyte trafficking, and zonation of metabolic functions[21]. Kupffer cells and other macrophage populations orchestrate inflammation and response to injury[22]. Gene therapies that target only hepatocytes may therefore be insufficient for diseases in which non-parenchymal cells are the primary drivers of pathology. Extending gene therapy beyond hepatocytes to these additional cell types is a key frontier[22].
Finally, the developmental context is crucial. Many of the severest inherited liver diseases present in infancy or early childhood, when early intervention could prevent irreversible damage. Importantly, pediatric livers are proliferative, which means that non-integrating vectors such as AAV are gradually diluted as hepatocytes divide, potentially leading to waning expression over time[3,23,24]. Conversely, integrating vectors or genome editing strategies that permanently modify the hepatocyte genome raise concerns about long-term safety and potential effects on growth and carcinogenesis or development of severe hepatocellular injury[25,26]. Thus, the liver’s biology offers powerful opportunities for gene therapy, but it also demands careful tailoring of approaches to disease type, stage, and patient age.
Delivery platforms for liver-directed gene therapy
The choice of delivery platform is crucial for the design of liver-directed gene therapies and significantly impacts their safety, efficacy, and durability. Among the available systems, hepatotropic AAV vectors, lentiviral vectors, and LNPs have emerged as the main options, each with distinct advantages and limitations that affect their suitability for various indications and patient populations. These established platforms, together with emerging approaches such as exosome-based systems, virus-like particles, and synthetic polymer nanocarriers, are compared in Figure 2, which summarizes their key advantages and limitations for liver targeting.

Figure 2. Comparison of different delivery platforms for liver-directed gene therapy. The figure highlights the main advantages and disadvantages of adenoviral vectors, adeno-associated virus (AAVs) vectors, lentiviral vectors, lipid nanoparticles (LNPs), exosomes, virus-like particles, and synthetic polymers/nanocarriers for hepatic gene transfer. Adenoviral vectors and AAVs are known for achieving high transduction efficiency in hepatocytes. Specific AAV serotypes like AAV8, AAV5, and AAV3B demonstrate strong liver tropism. However, they are constrained by immunogenicity, high seroprevalence of neutralizing antibodies, limited packaging capacity, and loss of episomal genomes in proliferating pediatric livers in the case of AAVs. Lentiviral vectors integrate into the host genome and can carry larger cassettes, enabling stable, long-term expression. This feature makes them suitable for ex vivo gene correction of hepatocytes or hepatocyte-like cells. Nevertheless, the risk of insertional mutagenesis and oncogenic activation currently restricts their direct in vivo liver applications. Non-viral systems, such as LNPs, exosomes, virus-like particles, and synthetic polymers/nanocarriers, have the ability to deliver mRNA, siRNA, and genome-editing components (such as CRISPR-Cas-based nucleases, base editors, and prime editors) with adaptable cargo size. They also offer the potential for repeated administration. However, they encounter challenges related to innate immune activation, reliance on LDLR-mediated uptake (for standard LNPs), cell-type specificity beyond hepatocytes, manufacturing complexity, and relatively limited clinical experience in systemic liver gene therapy.
AAV vectors have become the primary platform for in vivo liver-directed gene addition. Naturally occurring and engineered serotypes like AAV8, AAV5, and AAV3B exhibit strong tropism for hepatocytes following systemic administration[3,24]. Once delivered, AAV vector genomes typically persist as episomal concatemers in the nucleus, allowing for long-term transgene expression in non-dividing or slowly dividing cells while minimizing integration-related genotoxicity[24]. Clinical trials in hemophilia A and B utilizing liver-directed AAV gene therapy have shown sustained expression of factor VIII or IX over multiple years in numerous patients, resulting in significant reductions in bleeding episodes and factor concentrate usage[27]. Similar AAV-based approaches have progressed to clinical trials for monogenic metabolic liver diseases, such as ornithine transcarbamylase deficiency and Crigler-Najjar syndrome, with initial findings indicating biochemical correction in some participants[28,29].
However, the deployment of AAV vectors in the liver is constrained by several important factors. Many individuals harbor pre-existing neutralizing antibodies against common capsids, which can severely reduce vector transduction and preclude treatment[30,31]. Administration of high-dose AAV can induce robust humoral and cellular immune responses against the capsid and, occasionally, the transgene product[32]. Clinically, this often manifests as transient elevations in aminotransferases that are typically mitigated by corticosteroids, but in rare cases more serious hepatic or systemic toxicity has been observed[33]. The risk of immune responses and toxicity must be balanced against the need to deliver sufficient vector genomes per hepatocyte to achieve therapeutic transgene expression. Furthermore, AAV’s relatively small packaging capacity, on the order of 4.7 kb, limits the size of the cDNA and regulatory elements that can be delivered, complicating therapies for genes with large coding sequences or requiring complex regulatory architectures[32]. In pediatric patients, the non-integrating nature of AAV, which is an advantage from the perspective of genotoxicity, becomes a liability because vector genomes are diluted as hepatocytes proliferate, leading to decreased expression over time[32]. These challenges, immunogenicity, dose-related toxicity, and limited durability, have prompted extensive efforts to engineer next-generation AAV capsids.
Lentiviral vectors are another important class of delivery vehicles. These vectors integrate into the host genome, enabling stable and long-term expression even in proliferating cells. This feature is advantageous in pediatric settings or when sustained transgene production is required across many cell divisions[34]. In the context of liver disease, lentiviral vectors have primarily been used in ex vivo settings. Autologous hematopoietic stem cells have been transduced ex vivo to treat inherited metabolic diseases that secondarily affect the liver, illustrating the strength of lentiviral platforms in systemic disorders. Experimental approaches are exploring ex vivo gene correction of hepatocytes or hepatocyte-like cells derived from induced pluripotent stem cells, followed by transplantation into the liver. This leverages the regenerative capacity of the organ to expand corrected cells. Direct in vivo delivery of lentiviral vectors to the liver remains controversial due to concerns about insertional mutagenesis, oncogenic activation, and off-target integration in non-hepatic tissues. In particular, integration at certain genomic sites may promote hepatocarcinogenesis[34,35]. Although modern self-inactivating lentiviral vectors with improved integration profiles are significantly safer than earlier retroviral systems[36,37], the long-term risks in the context of chronic liver disease and potential pre-neoplastic changes require careful evaluation. For these reasons, lentiviral vectors currently have a more limited role in liver gene therapy, focusing on ex vivo applications and scenarios where the benefits of stable integration clearly outweigh the risks.
LNPs have quickly become a prominent non-viral platform for delivering nucleic acids to the liver due to their versatility[38]. These LNPs encapsulate various types of cargo, such as mRNA, small interfering RNA (siRNA), or CRISPR-Cas components, within a lipid shell made up of ionizable lipids, cholesterol, phospholipids, and polyethylene glycol-conjugated lipids[38]. Upon intravenous administration, LNPs are opsonized and interact with apolipoprotein E, which facilitates uptake by hepatocytes through low-density lipoprotein receptors, resulting in strong liver tropism[39]. In the clinical setting, LNP technology has been proven effective with approved siRNA therapeutics targeting genes expressed in hepatocytes and mRNA vaccines. While mRNA vaccines are not liver-specific, they have demonstrated the scalability and safety of the LNP platform. For gene therapy purposes, LNPs offer the advantage of delivering large cargos, such as mRNA encoding genome editors and guide RNAs, without integrating into the genome[38]. This allows for transient expression of potent genome-editing tools like base editors, which can induce permanent genetic changes while minimizing the risk of off-target effects[40]. Early clinical trials using LNP-formulated CRISPR-Cas9 components for in vivo genome editing in the liver have produced promising results. Significant knockdown of target genes has been achieved after a single infusion, demonstrating the feasibility of this approach.
Although LNPs efficiently deliver nucleic acids to hepatocytes through apolipoprotein E-mediated uptake pathways described above, achieving precise targeting of other hepatic cell populations remains challenging[41]. Efficient delivery to HSCs, cholangiocytes, or Kupffer cells will likely require additional engineering strategies, such as incorporating cell-specific ligands or modifying nanoparticle surface chemistry[41]. A promising lipid candidate (CL15A6) that allows effective mRNA delivery to activated HSCs by targeting clathrin-mediated endocytosis through the platelet-derived growth factor receptor-β showed high efficacy in a mouse model undergoing liver fibrosis[42]. Similarly, recently the incorporation of mannose-conjugated cholesterol allowed effective targeting of mRNA-dotted nanoparticles to liver sinusoidal endothelial cells and Kupffer cells following intravenous administration in mice[43].
Despite these strengths, LNP-based delivery also has limitations. Infusion-related reactions, triggered immune responses, and transient elevations in liver enzymes can occur, particularly at higher doses[40]. Achieving efficient editing or therapeutic expression at lower doses and increasing the transduction rates in patients lacking sufficient low-density lipoprotein receptor remains a priority, especially for indications requiring treatment in more fragile patients or in combination with other therapies[44]. Moreover, while hepatocyte targeting via apolipoprotein E is well established, extending precise targeting to other liver cell types, such as stellate cells, cholangiocytes, or Kupffer cells, will require additional engineering, for example by incorporating specific targeting ligands on the LNP surface[39]. Nonetheless, the modularity of LNPs, their favorable manufacturing profile, and the possibility of repeated dosing without eliciting long-lasting neutralizing antibodies make them highly attractive for many liver-directed applications.
Beyond these principal platforms, other modalities are emerging. Exosome-based delivery systems[45,46], virus-like particles[47], synthetic polymers/nanocarriers[48], and hybrid approaches[49] that combine viral and non-viral features are under investigation as potential alternatives or complements. They aim to improve cell-type specificity, reduce immunogenicity, and enable re-dosing. In parallel, advances in vector manufacturing, including suspension cell culture systems, improved purification methods, and continuous manufacturing processes, are gradually addressing scalability and cost barriers. Together, this diverse and rapidly evolving landscape of delivery platforms provides a versatile toolkit for tailoring liver gene therapies to specific disease contexts, patient characteristics, and therapeutic objectives.
Beyond safety considerations, therapeutic efficacy is a central challenge for liver-directed gene therapy. Achieving sufficient levels of hepatocyte transduction or editing is often necessary to reach the biochemical thresholds required for clinical benefit and reduce toxicities[50]. Some metabolic disorders exhibit favorable threshold effects, where restoration of a small fraction of normal enzyme activity can improve disease manifestations. However, many conditions require higher levels of correction or sustained expression across a substantial proportion of hepatocytes. Transduction efficiency can vary considerably between individuals due to differences in liver architecture and immune systems[24]. Factors such as fibrosis stage, vector pharmacokinetics, and host immune responses will impact gene transfer efficiencies[19,51]. Additionally, the heterogeneous distribution of vectors within the hepatic lobule may lead to uneven gene expression[50], potentially limiting therapeutic impact even when the total vector dose is high. Improving delivery efficiency while maintaining acceptable safety margins remains a major objective in the development of next-generation vectors and nanoparticle systems.
Therapeutic strategies: from gene addition to precision editing
Recent advances in genome editing technologies have expanded the therapeutic toolkit for liver diseases, building on earlier gene-addition approaches. This enables direct modification of endogenous loci in addition to conventional transgene delivery[52]. This gene-addition strategy remains highly relevant and forms the basis of many ongoing trials for monogenic metabolic liver diseases. In these treatments, an AAV vector carrying a cDNA controlled by a liver-specific promoter is delivered systemically, resulting in persistent episomal expression of the therapeutic protein. Since many metabolic diseases require only modest levels of transgene expression for biochemical correction, even a small amount of expression in some hepatocytes can significantly improve clinical outcomes. For example, restoring partial activity of urea cycle enzymes can prevent hyperammonemic crises, and low-level expression of UGT1A1 can decrease unconjugated bilirubin in Crigler-Najjar syndrome[53]. Gene addition is conceptually straightforward, typically produces a clear pharmacodynamic effect, and can be applied even when mutations are heterogeneous or distributed across large genes.
However, gene addition has limitations that become more evident as the field advances. This method does not modify the endogenous mutant locus, so it may not fully replicate the natural regulation of gene expression[54,55]. Transgene expression depends on the promoter and regulatory elements chosen, which may not match the timing, location, or amount of endogenous gene activity. Overexpression can cause cellular stress or toxicity, while underexpression may not provide enough benefit[54,55]. In diseases with dominant-negative or gain-of-function mechanisms, simply adding a wild-type allele may not be adequate and could, in some cases, worsen the disease. Additionally, in pediatric patients, non-integrating gene addition may require retreatment as the vector genomes are diluted with hepatocyte growth, but re-dosing is complicated by immune reactions to the vector.
These limitations have catalyzed a shift toward precision genome editing strategies that directly modify endogenous loci. CRISPR-Cas nucleases enable the targeted introduction of double-strand breaks at specific genomic sites, which are then repaired by cellular machinery through non-homologous end joining or homology-directed repair[56]. By delivering a Cas nuclease and a guide RNA to hepatocytes, one can disrupt pathogenic genes, correct point mutations, or insert therapeutic cassettes into safe harbors or endogenous loci. In the liver, CRISPR-Cas-mediated gene disruptions has been used in preclinical models to knock out genes that promote disease, such as those involved in cholesterol synthesis or viral replication[56]. More sophisticated strategies exploit homology-directed repair or homology-independent targeted integration to insert corrective sequences into precise genomic locations, restoring physiological regulation.
However, double-strand break-based editing raises concerns about genotoxicity, including large deletions, chromosomal rearrangements, and activation of p53 pathways[57,58]. This has led to the development of editing modalities that do not rely on double-strand breaks. For example, base editors combine a catalytically impaired Cas protein with a deaminase enzyme, allowing for targeted conversion of single nucleotides (such as C to T or A to G) within a specific window. This enables the correction of many pathogenic point mutations or the introduction of protective variants in regulatory elements while minimizing the risk of large-scale genomic rearrangements. Prime editors further expand the range of possible edits by utilizing a reverse transcriptase fused to Cas and a specialized guide RNA (a prime editing guide RNA) that encodes the desired change[59]. Prime editing can introduce small insertions, deletions, or arbitrary point mutations without the need for donor templates or double-strand breaks, although its efficiency in vivo is still being optimized[59,60].
Another important factor influencing therapeutic success is the efficiency of genome editing within target hepatocytes. While preclinical models often show high editing rates under controlled experimental conditions, achieving similar efficiencies in humans is much more difficult[61]. Editing outcomes are influenced by various factors, such as vector delivery efficiency, availability of editing components within cells, chromatin accessibility at the target site, and cellular DNA repair pathways. In many diseases, only a portion of hepatocytes may be edited in vivo, leading to questions about whether the correction levels achieved are adequate for lasting clinical benefits. Current strategies that are being investigated to overcome this limitation include improved guide RNA design, optimized editor variants with increased catalytic activity, and delivery methods that temporarily boost intracellular levels of editing machinery while minimizing prolonged exposure that could raise the risk of off-target effects[62].
These tools are particularly well-suited for liver applications, where specific point mutations underlie many inherited diseases, and where precise tuning of gene expression through regulatory edits could modulate complex conditions[63,64].
An additional strategic evolution involves expanding therapeutic targets beyond protein-coding sequences to include promoters, enhancers, splicing junctions, and noncoding RNA[65]. Regulatory regions that control gene expression, such as enhancers and promoters, as well as non-coding RNAs that modulate hepatic metabolism and fibrosis are increasingly recognized as attractive intervention points. Editing or epigenetically reprogramming these elements offers a way to adjust the expression of endogenous genes in a graded fashion, potentially yielding more physiological and durable effects than overexpression from exogenous cassettes. For example, editing transcription factor binding sites in the promoter of a key metabolic enzyme could moderately upregulate its expression in response to endogenous signals, while targeting long non-coding RNAs involved in stellate cell activation might attenuate fibrogenesis without completely abolishing necessary wound-healing responses[65].
Another conceptual shift involves cell-type specificity. To date, most liver gene therapy approaches have primarily focused on hepatocytes. However, there is now increasing attention being directed towards non-parenchymal cell populations involved in fibrosis, inflammation, and cholestatic disease[20,66]. Emerging work is beginning to target these other cell types. For instance, therapies directed at stellate cells aim to reverse or halt fibrosis by downregulating profibrotic genes, upregulating antifibrotic mediators, or reprogramming activated stellate cells back to a quiescent phenotype[66]. Cholangiocyte-targeted approaches are being explored for genetic cholangiopathies and cholestatic conditions, where correcting transporter function or modulating bile acid signaling in the biliary epithelium could have profound effects on disease progression[67]. Immune cell-directed therapies, including liver-resident macrophages and (innate-like) T cells, may modulate chronic inflammation and autoimmunity or enhance anti-tumor responses in HCC[68].
Ultimately, therapeutic strategies for liver disease are likely to become increasingly nuanced and multimodal. In some cases, such as simple loss-of-function monogenic metabolic disorders, traditional hepatocyte-directed gene addition may remain the most practical approach, especially if started early before significant tissue damage occurs. In other situations, such as dominant disorders, complex polygenic diseases, or advanced fibrosis, precision editing and cell-type-specific targeting will be necessary to achieve meaningful and long-lasting benefits. Genome editing can also be combined with temporary modulation of pathways using RNA-based therapies, small molecules, or biologics. The emerging toolkit of gene addition, knockout, knock-in, base editing, prime editing, and epigenome editing, along with the ability to select among hepatocytes and various non-parenchymal cells as targets, provides a foundation for a flexible, programmable therapeutic toolkit that can be customized to the underlying mechanism of different liver diseases.
DISEASE INDICATION LANDSCAPE: LESSONS FROM EARLY CLINICAL EXPERIENCE
The development of liver-directed gene therapy has been influenced by the choice of initial disease indications. Rare monogenic metabolic liver diseases were identified early as ideal candidates due to their severity, lack of effective long-term treatments, and clear biochemical and genetic endpoints that allow for monitoring therapeutic benefit. Clinical trials in these conditions have provided valuable insights into dose-response relationships, the longevity of expression, immune responses, and the impact of disease stage and patient age on outcomes.
In urea cycle disorders, such as ornithine transcarbamylase deficiency, AAV-mediated gene addition aims to restore enzymatic activity in hepatocytes to prevent hyperammonemic crises and associated neurological damage in preclinical models and human studies[69-71]. Early-phase trials have shown that vector delivery can result in detectable transgene expression and reduced ammonia levels in some participants, particularly in adults or adolescents with residual liver function[70]. However, maintaining therapeutic enzyme levels over time has proven to be challenging, especially in younger patients. This difficulty likely stems from both vector dilution in a growing liver and immune-mediated loss of transduced cells. These experiences emphasize the importance of carefully selecting the timing of intervention and underscore the need for strategies that allow re-dosing or more lasting correction in pediatric populations.
Crigler-Najjar syndrome, characterized by a deficiency in the bilirubin-conjugating enzyme UGT1A1, provides another instructive example[72]. Patients with the severe form of the disease develop significant unconjugated hyperbilirubinemia and are at risk for kernicterus, often necessitating intensive phototherapy and, in many cases, liver transplantation. AAV-based gene addition trials have shown that restoring UGT1A1 expression in hepatocytes can lower serum bilirubin levels and reduce the need for phototherapy in some treated individuals[53]. However, both the magnitude and durability of the response have varied, with some patients experiencing only partial or temporary benefits. The outcomes of these trials demonstrate how even small improvements can have clinical significance, while also highlighting the importance of maintaining transgene expression over time, especially in conditions where the continuous production of a harmful metabolite poses a lifelong threat.
Other monogenic metabolic disorders, such as glycogen storage diseases, Wilson disease, and certain fatty acid oxidation defects, are currently undergoing active preclinical and early clinical exploration[73-76]. In glycogen storage disease type Ia, for example, gene addition is being used to restore glucose-6-phosphatase activity, with the goal of correcting hypoglycemia and lactic acidosis. Similarly, in Wilson disease, gene therapies are being developed to restore ATP7B function and normalize copper homeostasis. Together, these programs support the concept that the liver can serve as a central therapeutic target for systemic metabolic diseases. However, they also emphasize the importance of understanding the specific pathophysiology of each disease. In cases where extrahepatic manifestations, such as neuromuscular involvement or cardiac complications, are significant, liver-directed therapies may need to be combined with interventions targeting other tissues.
Cholestatic and biliary diseases pose unique challenges. Progressive familial intrahepatic cholestasis (PFIC), caused by mutations in genes encoding bile transporters and other biliary proteins, is a group of monogenic disorders. In PFIC, cholestasis results in pruritus, growth failure, and progressive liver damage. Gene therapy strategies for PFIC face the challenge that, depending on the subtype, the primary defect may be in hepatocytes or cholangiocytes[77,78]. Correcting transporter function in hepatocytes, such as restoring bile salt export pump (BSEP) or multidrug resistance protein 3 (MDR3) activity, could normalize bile flow and reduce toxicity, but achieving sufficient expression in the appropriate membrane domains is complex[77,78]. Additionally, many patients present with advanced fibrosis or cirrhosis, where structural changes in the liver and biliary tree may limit the reversibility of the disease even if the underlying defect is corrected. Biliary atresia, a leading cause of pediatric liver transplantation, presents even greater complexity as it involves inflammatory and fibrotic obliteration of the extrahepatic bile ducts. The role of gene therapy in biliary atresia may be more limited or may involve immunomodulatory approaches combined with surgical interventions to preserve or reconstruct bile flow.
HCC introduces additional complexity to the disease indication landscape. HCC often arises in the context of chronic liver disease and cirrhosis, where the surrounding liver parenchyma is already compromised. Gene therapy strategies for HCC must address tumor heterogeneity and both intra-tumoral and inter-tumoral genetic diversity. Moreover, there is a dual need to target malignant cells while sparing normal hepatocytes. Approaches under investigation include oncolytic viruses that selectively replicate in tumor cells, vectors delivering suicide genes or prodrug-converting enzymes, and gene therapies that enhance anti-tumor immunity, such as local expression of cytokines or checkpoint inhibitors[79-81]. Additionally, cell therapies, including chimeric antigen receptor (CAR) T cells engineered to recognize liver tumor antigens, may be considered part of the same broader category of gene-based interventions for liver cancer[82].
In contrast to inherited metabolic liver disorders, gene delivery to HCC presents additional biological challenges. Tumor tissues are characterized by pronounced cellular heterogeneity, abnormal vascular architecture, and a complex stromal microenvironment that can impede vector penetration and uniform distribution[79]. Moreover, malignant hepatocytes frequently display altered receptor expression and metabolic states that may influence vector uptake and intracellular processing. As a result, delivery systems optimized for normal hepatocytes may not achieve comparable efficiency in tumor cells. An additional complication is that many patients with HCC develop tumors in the setting of advanced fibrosis or cirrhosis. Cirrhotic livers are characterized by profound architectural remodeling, including fibrotic septa, capillarization of sinusoids, altered hepatic blood flow, and increased extracellular matrix deposition[83]. These structural changes can create stromal barriers that restrict vector penetration and reduce the uniform distribution of therapeutic agents within tumor tissue. As a result, delivery systems optimized in healthy liver models may exhibit substantially lower efficiency in cirrhotic tumor-bearing livers, highlighting the need for delivery strategies specifically adapted to the pathological microenvironment of HCC.
Several strategies are currently being explored to improve tumor-selective targeting. These include the use of tumor‑specific promoters, engineered oncolytic viruses that preferentially replicate in malignant cells, and vectors displaying ligands for receptors enriched in tumor tissue[79]. In parallel, gene-modified immune cell therapies such as CAR-T or TCR-engineered lymphocytes are being investigated as complementary approaches to target liver tumors through immune-mediated mechanisms[82].
To place these individual programs and strategies in a broader translational context, Table 2 summarizes the current development stages of gene therapy approaches across major groups of liver-related diseases, including the dominant modalities, target cell types, and highest clinical phases reached to date.
Overview of gene therapy development stages for liver-related diseases
| Disease group/category | Representative indications (examples) | Main gene-therapy modality/target cells | Highest clinical stage | Example targets/vectors | Key notes (primary liver disease vs. systemic, etc.) |
| Coagulation disorders treated via liver | Hemophilia A (F8) and Hemophilia B (F9) | In vivo AAV gene addition to hepatocytes[84-86] | Approved/Phase III | AAV5F8, AAV5F9 (Padua) and other AAV serotypes | In these disorders, the liver functions as a “protein factory” that produces therapeutic clotting factors, even though the underlying disease does not primarily affect the liver. Several therapies have been approved, including Roctavian, Hemgenix, and Beqvez |
| Urea cycle disorders | Ornithine transcarbamylase (OTC) deficiency | In vivo AAV gene addition to hepatocytes[28,70] | Phase I/II (some planning pivotal) | AAVOTC constructs with liverspecific promoters | Primary metabolic liver disease is the focus of multiple first-in-human trials with an emphasis on controlling ammonia levels and ensuring safety |
| Glycogen storage disease type I (GSD Ia) | G6PC deficiency | In vivo AAV gene addition to liver[73,74] | Phase I/II, pivotal planning | AAVG6PC vectors | Early clinical studies have shown improved fasting tolerance in patients |
| Other glycogen storage diseases | e.g., GSD III and other disorders | AAV gene addition, sometimes mRNA/LNP[87] | Preclinical and early Phase I | AAV-based vectors or LNP mRNA enzymes related to glycogen | Most programs remain at the animal-model stage, with only a few early first-in-human studies |
| Organic acidemias | Methylmalonic acidemia (MMA) and Propionic acidemia (PA) | In vivo AAV gene addition to liver (with or without muscle)[88,89] | Phase I/II | AAV-MUT (MMA), AAV-PCCA/PCCB (PA) | Primary metabolic liver disease with systemic manifestations is being studied |
| Crigler–Najjar syndrome type I | UGT1A1 deficiency | In vivo AAV gene addition to hepatocytes[29,53] | Phase I/II | AAV-UGT1A1 | This is a rare bilirubin conjugation defect, and some patients show reduced bilirubin levels requiring less phototherapy. However, concerns remain about the durability and liver inflammation at high doses |
| Progressive familial intrahepatic cholestasis (PFIC) | PFIC subtypes (e.g., BSEP/PFIC2, MDR3/PFIC3) | AAV gene addition and gene editing under exploration[77,78] | Preclinical and early Phase I/II | AAV-ABCB11, AAV-ABCB4, etc. | Primary cholestatic liver diseases are the focus of most programs, with most still in the preclinical stage, and a few early clinical trials measuring bile acids and liver function |
| Wilson disease | ATP7B mutations (copper transport) | In vivo AAV gene addition (fulllength or mini-ATP7B)[75,76] | Phase I/II | AAV-ATP7B variants | A primary liver/neurologic disease has initiated early human trials focusing on copper handling and liver function |
| Alpha1 antitrypsin deficiency (A1ATD) | SERPINA1 (Z allele) related liver and lung disease | AAV gene addition; gene editing/knockdown of mutant A1AT[90] | Preclinical and early Phase I | AAV-SERPINA1 and RNAi editing targeting mutant A1AT in hepatocyte | For a dual liver and lung disease. Most gene therapy work is still in the preclinical stage or just entering first-in-human studies |
| Other inherited metabolic liver disorders | Various ultra-rare enzyme or transporter defects (bile acid synthesis defects, rare transporters, etc.) | AAV gene addition and early editing approaches | Mostly preclinical; a few Phase I | Disease-specific AAV constructs | Many ultra-rare conditions have limited programs with animal data. Some highly specialized first-in-human trials exist but are small and in the early stages |
| Chronic hepatitis B (HBV) | Chronic HBV infection (efforts for functional cure) | In vivo gene editing (e.g., CRISPR, TALENs) targeting HBV DNA/cccDNA and also gene silencing methods (siRNA/ASO)[91] | Phase I/II for gene editing; Phase II/III for RNAi | CRISPR-Cas systems, other nucleases, and various siRNA/ASO drugs | The liver is the primary site of infection, and current gene editing programs targeting viral hepatitis remain at an early stage and primarily focus on safety. RNAi/ASO drugs are more advanced but are not typically considered “gene therapy” in the strict sense |
| Other viral hepatitis (HCV, HDV, etc.) | HCV and HBV/HDV co-infection, etc. | Limited focus on genetherapy and mostly usage of small molecules/biologics | No major liver-directed gene therapy beyond exploratory or preclinical | Not applicable for routine clinical trials | HCV is effectively treated with DAAs, so gene therapy is not a major clinical focus. Gene-modified immune cells (e.g., for HIV/HBV) are more active than liver-targeted vectors |
| Common chronic liver diseases | MASH/MASLD, cirrhosis (various causes), autoimmune hepatitis, PBC, PSC | Antifibrotic or immune-modulating gene transfer, targeting of genes associated with fat metabolism, editing of fibrogenic pathways[92-94] | Mostly preclinical and some early Phase I studies | AAV or non-viral vectors targeting hepatocytes or hepatic stellate cells (experimental) | Primary chronic liver diseases are dominated by small molecules and biologics in clinical development. Gene therapy remains experimental, mostly in animal models |
| Liver cancers | HCC and intrahepatic CCA | Oncolytic (e.g., VG161) and gene-armed viruses, as well as gene-modified cell therapies (CART/TCRT)[81,82] | Phase I/II | Adenoviral, poxviral, and other oncolytic vectors; CAR-T/TCR-T against HCC antigens | There are numerous early trials globally, with some oncolytic/gene therapies approved for other cancers in certain countries, but there is no widely adopted gene therapy standard of care for HCC/CCA |
| Systemic diseases treated via liver-targeted gene therapy | ATTR amyloidosis, familial hypercholesterolemia, and some complement disorders | In vivo CRISPR/base-editing or AAV in hepatocytes (to silence or correct systemic targets)[64] | Phase I/II | CRISPR editing (of TTR), base editing (of PCSK9, ANGPTL3), and AAV gene addition for systemic proteins | The liver is the therapeutic target organ, but diseases primarily affect the heart, nerves, vasculature, etc. These programs showcase mature liver-targeting technology that is expected to influence future liverdisease therapies |
These strategies illustrate how gene therapy in the liver can move beyond correction of inherited metabolic defects to address malignant transformation and immune evasion, but they also highlight complex safety considerations, including the risk of exacerbating underlying liver dysfunction and provoking severe inflammatory responses.
Beyond individual indications, early clinical experiences across diseases have yielded several overarching lessons. First, the timing of intervention relative to disease progression is critical. In conditions where irreversible fibrosis or architectural distortion has developed, correcting the primary molecular defect may not fully restore function. However, early treatment may prevent cumulative damage but must contend with the practical and ethical challenges of intervening in infants or even in utero. Second, inter-individual variability in vector pharmacokinetics, hepatocyte transduction efficiency, and immune responses is substantial and not yet fully predictable. This underscores the need for biomarkers and genomic or immunologic profiling that can guide personalized dosing and risk stratification. Third, genotype-phenotype correlations can inform patient selection. For example, individuals with residual enzyme activity or milder mutations may benefit from lower levels of transgene expression, while those with severe null mutations may require more robust correction.
Collectively, these experiences suggest that liver-directed gene therapy is most likely to succeed when disease mechanisms are well understood, clear and sensitive biomarkers of efficacy are available, and interventions are timed to maximize the potential for functional restoration. They also point toward a future in which liver gene therapies will be deployed across a spectrum of indications, from rare monogenic diseases to common multifactorial conditions and malignancies, but with approaches tailored to the specific biological and clinical context of each disease.
Key challenges and safety considerations: a personal view
The translation of liver-directed gene therapy from preclinical models to clinical practice has highlighted several challenges that must be addressed to fully realize its potential. Immune responses to vectors and transgene products, dose-dependent toxicities, risks of genotoxicity and oncogenesis, durability of effect in the context of liver growth and regeneration, as well as practical issues related to manufacturing, cost, and equitable access are all central concerns.
Immunogenicity remains a major barrier to liver-directed gene therapy. As described ealier, AAV vectors can be affected by preexisting neutralizing antibodies, immune responses against capsid proteins, and limitations in vector re-administration. These immune mechanisms can reduce hepatocyte transduction efficiency and may lead to transient elevations in liver enzymes or loss of transgene expression. Consequently, strategies such as capsid engineering, transient immunomodulation, and alternative delivery platforms are being explored to mitigate immune responses and enable safer and more durable therapeutic outcomes[95]. Cellular immune responses, especially CD8+ T cells targeting capsid-derived peptides presented by hepatocytes, can result in temporary or prolonged loss of transgene-expressing cells and elevated liver enzymes. This may require immunosuppressive therapy to maintain transgene expression and prevent hepatitis.
A practical limitation is the difficulty of vector re-administration. Following exposure to AAV capsids, most patients develop durable neutralizing antibodies that prevent effective redosing with the same or closely related serotypes[52]. This constraint is particularly relevant for pediatric patients and chronic conditions in which therapeutic expression may decline over time. Current research therefore focuses on capsid engineering, transient immune modulation, and the use of alternative delivery systems that may allow repeated administration.
Similar concerns, albeit with different timing and mechanisms, apply to non-viral platforms such as LNPs. Innate immune activation and complement activation-related pseudoallergy can occur, especially at higher doses[96,97]. Strategies to address these immune responses include engineering capsids to reduce recognition by pre-existing antibodies, temporary immunosuppression during and after vector administration, and exploring alternative platforms including LNPs or engineered exosomes that may allow for repeated dosing. However, the inability to easily re-dose many current vectors is a significant limitation, particularly for pediatric applications and chronic diseases.
Toxicity and genotoxicity are closely related concerns. As discussed, high-dose systemic administration of AAV vectors has been associated with acute hepatotoxicity, thrombocytopenia, microangiopathy, and, in rare cases, severe liver injury or multi-organ failure. The exact mechanisms remain incompletely understood but likely involve a combination of innate immune activation, complement activation, direct effects of capsid proteins, and possible off-target transduction of other tissues. Clinical experience has shown that administering very high systemic doses of AAV vectors can lead to significant toxicity[98]. This toxicity may include acute hepatocellular injury, thrombocytopenia, complement activation, and, in rare cases, thrombotic microangiopathy. These adverse events are primarily observed in trials that require high vector genome doses to achieve effective hepatocyte transduction[99]. This underscores the narrow therapeutic window that limits many AAV-based liver gene therapies. Therefore, improving vector potency and hepatocyte tropism to reduce the total vector dose required for therapeutic efficacy has become a primary objective of current vector engineering efforts.
Vigilant monitoring and dose titration in clinical trials are essential. In the long term, although AAV is mostly non-integrating, low-level integration events can occur. In animal models, AAV integration events have occasionally been associated with HCC, especially when vectors were administered at very high doses in neonatal animals or in the presence of underlying oncogenic stress. The extent to which these findings apply to humans is still being actively researched. Careful analysis of integration site and long-term monitoring of treated patients are crucial to evaluate any potential increased risk of malignancy.
Genome editing introduces additional safety considerations beyond those associated with gene addition. As discussed in the section on precision editing strategies, double-strand break-based approaches such as CRISPR-Cas nucleases can generate unintended genomic alterations, including large deletions or chromosomal rearrangements[57,100]. These effects may include potential deamination at off-target sites or unintended edits in sequences with partial homology to the target. Although newer tools such as base editors and prime editors reduce reliance on double-strand breaks, careful assessment of off-target activity and long-term genomic stability remains essential. Delivering editor components through high-dose AAV or LNPs can introduce issues of immunogenicity and toxicity. To address these risks, thorough preclinical assessment of off-target editing sites using unbiased genome-wide methods is essential. Dose optimization and transient expression strategies may help minimize exposure to editing components. In addition, post-treatment genomic surveillance may be required. This becomes especially crucial when these technologies are used in younger patients with longer lifespans, as adverse effects may become more apparent over time.
Durability of therapeutic effects is another key challenge, particularly in pediatric populations. As mentioned earlier, hepatocyte proliferation during liver growth can dilute nonintegrating vector genomes such as AAV episomes, leading to a gradual loss of transgene expression[25,52]. Clinical experience in hemophilia and metabolic diseases suggests that expression levels achieved with AAV can remain relatively stable in adults over several years but may decline more rapidly in younger patients[85,52]. Integrating vectors or genome editing strategies that permanently modify the hepatocyte genome can overcome this limitation, but at the expense of increased concern about long-term genomic effects and oncogenic potential. This trade-off highlights the need for age-tailored strategies: for example, using transient gene addition or supportive therapies to bridge young children to an age where more durable interventions can be safely administered, or developing re-dosing protocols that utilize different serotypes or platforms to circumvent neutralizing antibodies. Observations from long-term clinical follow-up studies further emphasize the importance of sustained therapeutic durability. In several gene therapy trials, especially in hemophilia, transgene expression has been detectable for multiple years but has shown gradual decline in some patients over time[101]. The mechanisms underlying this variability remain incompletely understood and may involve immune-mediated loss of transduced hepatocytes, epigenetic silencing of vector genomes, or hepatocyte turnover. These findings underscore the importance of developing strategies that support sustained expression, enable vector re-administration, or achieve permanent genomic correction through editing-based approaches.
Beyond biological and clinical challenges, practical and societal issues also play a significant role. Manufacturing AAV vectors and LNPs at scale with consistent quality is a technically demanding task. Production is often hindered by low yields, limited scalability, and analytical complexity. These challenges result in high production costs and uncertainties regarding long-term durability and safety[52,102,103]. The capacity of current production infrastructure may be insufficient to meet the potential demand if gene therapies are to be widely adopted for common liver diseases, not just rare monogenic conditions. High production costs, combined with the complexity of delivery and long-term follow-up, translate into expensive therapies that raise questions about affordability and equitable access. These concerns are particularly acute for liver diseases that are highly prevalent in low- and middle-income countries, such as viral hepatitis-related cirrhosis and HCC, where healthcare resources are limited. Addressing these disparities will require not only advances in manufacturing technology to reduce costs but also novel pricing models, global partnerships, and policies that prioritize equitable distribution. In addition to biological safety concerns, several practical challenges have become apparent during clinical translation. Patient eligibility can be limited by the presence of pre-existing neutralizing antibodies against common AAV capsids, which may exclude a substantial proportion of the population from treatment. Furthermore, manufacturing clinical-grade viral vectors at large scale remains technically demanding and costly, potentially limiting broad access if these therapies are expanded beyond rare diseases. Recent work has suggested that machine learning and advances in viral immunology can enable engineering of a new generation of de-immunized capsids beyond the antigenic landscape of natural AAVs[104]. Long-term clinical monitoring is also required to evaluate durability of expression and potential late adverse effects, adding additional logistical complexity to the implementation of gene therapy in routine clinical care.
Finally, ethical considerations are pervasive in the field of gene therapy, especially when discussing germline genome editing for inherited diseases[105]. When it comes to treating children, especially using irreversible genome editing, there must be a delicate balance between the potential to prevent severe, life-limiting diseases and the uncertainties surrounding long-term risks[106]. In utero interventions, which are being considered for certain severe diseases, bring up additional ethical dilemmas regarding consent, risks to the fetus and the pregnant individual, and the possibility of unintended consequences on future generations, even if germline modification is not the goal. Open and honest communication with patients and their families about the benefits and risks, strong regulatory oversight, the establishment of long-term registries to monitor outcomes, and inclusive discussions involving patient advocacy groups, ethicists, and diverse communities are all crucial to ensure that advancements in liver gene therapy are carried out responsibly.
Future directions: toward programmable and regenerative liver therapies
It is important to distinguish between technologies that have already demonstrated clinical efficacy and those that remain at an experimental or early translational stage. Liver-directed AAV gene addition therapies for hemophilia and certain systemic disorders represent the most clinically advanced applications and have reached late-phase trials or regulatory approval in some jurisdictions[4,107]. By contrast, many genome-editing strategies, including base editing, prime editing, and epigenome editing, are still largely in preclinical development or early clinical testing. Similarly, approaches targeting non-parenchymal liver cells or complex polygenic diseases remain largely conceptual. Recognizing these differences in maturity is critical when evaluating the near-term clinical potential of emerging gene therapy platforms.
Looking ahead, the field of liver-directed gene therapy is evolving from a collection of disease-specific products to a more integrated ecosystem of programmable platforms that can be adapted across indications and combined with regenerative and immunomodulatory strategies. Several converging research directions are likely to shape this future. One central theme is the development of next-generation vectors and delivery systems that improve specificity, potency, and safety. Advances in protein engineering, high-throughput screening, and machine learning are being leveraged to create AAV capsids with enhanced hepatocyte tropism, reduced binding to pre-existing antibodies, and minimized off-target transduction of other tissues such as the dorsal root ganglia or myocardium[108,109]. Directed evolution approaches, performed in vivo in animal models or using humanized liver chimeric mice, can identify capsids that efficiently transduce human hepatocytes. These strategies can yield capsids that efficiently transduce human hepatocytes at lower doses, thereby reducing toxicity and manufacturing demands. Parallel efforts aim to engineer LNPs with tailored properties, including optimized ionizable lipids, surface modifications with targeting ligands, and controlled release characteristics. These improvements may enable preferential delivery to specific liver cell types such as HSCs or cholangiocytes. Such precision delivery systems could form the backbone of modular therapies, in which the therapeutic cargo (for example, a base editor or regulatory RNA) can be exchanged while the delivery platform remains largely unchanged.
A second theme involves the refinement of genome editing to achieve safer, more versatile, and more predictable outcomes. The trajectory of editing tools is moving away from double-strand break-dependent systems toward approaches that minimize DNA damage. Next-generation base editors with reduced off-target deamination and improved specificity, as well as prime editors with enhanced efficiency, are actively being optimized for in vivo use[110]. RNA editing platforms, particularly antisense RNA-guided adenosine deaminase acting on RNA (ADAR) which operate at the transcript level and thus do not permanently alter the genome, provide another layer of flexibility, particularly for conditions where reversible modulation of gene expression or function is advantageous[111]. Additionally, epigenome editing tools that recruit transcriptional activators or repressors to specific loci without changing the underlying DNA sequence offer a way to titrate gene expression up or down, which may be especially useful in polygenic or multifactorial liver diseases where subtle modulation of multiple pathways may be preferable to complete ablation or overexpression of a single target[112].
A third area of active development involves harnessing and directing the regenerative capacity of the liver. Gene therapy can intersect with regenerative medicine in several ways. In situ reprogramming strategies aim to convert one cell type into another within the liver, for example by reprogramming activated stellate cells or myofibroblasts into hepatocyte-like cells, thereby simultaneously reducing fibrosis and increasing functional parenchyma[113]. Gene therapies can also be used to enhance the engraftment and proliferation of transplanted gene-corrected cells, such as hepatocytes or liver organoids derived from patient-specific induced pluripotent stem cells[114]. In this scenario, patients might receive a combination of ex vivo gene-corrected cell transplants and in vivo gene therapies that modulate the microenvironment to favor engraftment, survival, and appropriate integration of these cells into the liver architecture. Partial reprogramming approaches that transiently activate developmental transcriptional programs or transcription factors without fully reverting cells to a pluripotent state are being explored as a means to stimulate regeneration while minimizing oncogenic risk, and gene therapy tools could provide temporal and spatial control over such interventions[115].
Combination therapies are likely to become increasingly important, particularly for complex or advanced liver diseases. For example, in patients with HCC arising in cirrhotic livers, gene therapies may directly target tumor cells, example through oncolytic viruses or constructs delivering pro-apoptotic genes. They may be combined with systemic immunotherapies, such as checkpoint inhibitors, as well as anti-fibrotic gene therapies directed at HSCs. In chronic viral hepatitis, gene editing designed to disable viral genomes or host factors essential for viral replication could complement antiviral drugs and immunomodulatory treatments, potentially leading to deeper and more durable cures. In metabolic diseases, gene addition or editing might be paired with small molecules that modulate metabolic pathways or with diet-based interventions, allowing lower doses of gene therapy and reducing toxicity. Rational design of such multimodal regimens will require systems-level understanding of liver pathophysiology and careful clinical trial design to assess synergistic effects and manage overlapping toxicities.
Data-driven personalization will also shape future liver gene therapies. High-resolution multi-omics profiling of liver tissue and blood, including genomics, transcriptomics, proteomics, and metabolomics, can provide detailed snapshots of disease state, treatment response, and emerging resistance mechanisms[116,117]. These tools can be combined with advanced imaging and non-invasive biomarkers such as circulating cell-free DNA and extracellular vesicles. Together, they may enable the dynamic adjustment of gene therapy strategies over time. For example, baseline genomic data might identify patients at higher risk for off-target editing or vector integration at specific hotspots, guiding vector choice or the intensity of post-treatment surveillance[117]. Longitudinal monitoring could detect early signs of clonal expansion or pre-neoplastic changes, prompting interventions or closer follow-up. Artificial intelligence-driven analysis of large datasets from gene therapy registries and trials could uncover patterns that inform dosing, vector selection, and identification of subgroups that derive particular benefit or experience specific adverse events[118,119].
Finally, the future of liver gene therapy will be shaped by how effectively the field addresses ethical, regulatory, and societal considerations[120]. Ensuring that advances do not exacerbate existing health disparities will require concerted efforts to make therapies affordable and accessible, including in regions with high burdens of liver disease. Engagement with diverse patient communities and stakeholders is fundamental to building trust, understanding preferences and concerns, and co-designing interventions and follow-up protocols. Regulatory frameworks will need to balance the urgency of addressing severe, otherwise untreatable diseases with the caution warranted by irreversible interventions with long-term unknowns. Innovative trial designs in clinical research, such as adaptive trials and platform trials integrated in Integrated Research Platforms, may be particularly well suited to evaluate modular gene therapy across multiple diseases[121]. Long-term registries and international collaborations will be essential to capture late-emerging outcomes and to refine best practices over time. Consequently, the future of liver-directed gene therapy probably lies in more precise, programmable, and context-specific interventions that combine gene addition, editing, and reprogramming with pharmacologic, cellular, and immunologic therapies. Achieving this vision will require not only technological advances but also careful clinical translation, thorough safety monitoring, and a dedication to fair implementation.
CONCLUSIONS
Liver-directed gene therapy has evolved from a theoretical concept to a rapidly advancing clinical reality with the potential to revolutionize the outcomes of severe liver diseases. The anatomical accessibility of the liver, its central physiological role, and its regenerative capacity make it an attractive target for therapeutic intervention. Early clinical trials in monogenic metabolic and cholestatic disorders demonstrate that restoring even limited hepatocyte function can produce meaningful clinical benefit. However, these trials have also highlighted the challenges of translating gene-based therapies into safe, effective, and widely available treatments. Issues such as immune reactions to vectors, dose-related side effects, concerns about genetic damage or cancer development, difficulties in treating children with growing livers, and the high costs and technical complexities of manufacturing these therapies all present significant hurdles to overcome. Recent advances in delivery systems, genome editing technologies, and high-resolution genomics are accelerating the clinical development of liver-directed gene therapies. AAV vectors, lentiviral systems, and LNPs each offer complementary strengths, while CRISPR-Cas, base editing, and prime editing enable increasingly precise and versatile manipulation of the hepatic genome.
Moving beyond gene addition in hepatocytes, emerging strategies target non-parenchymal cells, modulate regulatory elements and non-coding RNAs, and intersect with regenerative and immunotherapeutic approaches. The conceptual shift from fixed, single-disease products to modular, programmable platforms holds the promise of more efficient development and broader applicability across a spectrum of liver conditions, from rare inherited diseases to common multifactorial disorders and malignancies. Translating liver gene therapy into routine clinical care will require close collaboration between basic scientists, clinicians, engineers, regulators, and patient communities. It will demand rigorous preclinical evaluation, cautious and transparent clinical translation, robust long-term follow-up, and proactive strategies to address issues of access and equity. If these translational barriers can be overcome, gene therapy may increasingly complement established treatments such as pharmacotherapy and liver transplantation. For selected conditions, it could substantially modify the long-term course of liver disease.
DECLARATIONS
Acknowledgments
The graphical abstract illustration and the two figures of this perspective were created using Servier Medical Art (SMART) and are licensed under the Creative Commons Attribution 4.0 International License (CC BY 4.0).
Authors’ contributions
The author contributed solely to the article.
Availability of data and materials
Not applicable.
AI and AI-assisted tools Statement
During the preparation of this manuscript, the AI tool EditMyEnglish (https://www.editmyenglish.com/) was used solely for language editing. The tool did not influence the study design, data collection, analysis, interpretation, or the scientific content of the work. The author takes full responsibility for the accuracy, integrity, and final content of the manuscript.
Financial support and sponsorship
None.
Conflicts of interest
The author serves as a Section Editor for Journal of Translational Genetics and Genomics. He was not involved in any aspect of the editorial handling of this manuscript, including reviewer selection, manuscript processing, or editorial decision-making.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Kostallari E, Schwabe RF, Guillot A. Inflammation and immunity in liver homeostasis and disease: a nexus of hepatocytes, nonparenchymal cells and immune cells. Cell Mol Immunol. 2025;22:1205-25.
2. Karlsen TH, Lammert F, Thompson RJ. Genetics of liver disease: from pathophysiology to clinical practice. J Hepatol. 2015;62:S6-S14.
3. Mücke MM, Fong S, Foster GR, Lillicrap D, Miesbach W, Zeuzem S. Adeno-associated viruses for gene therapy - clinical implications and liver-related complications, a guide for hepatologists. J Hepatol. 2024;80:352-61.
4. Pierce GF, Fong S, Long BR, Kaczmarek R. Deciphering conundrums of adeno-associated virus liver-directed gene therapy: focus on hemophilia. J Thromb Haemost. 2024;22:1263-89.
5. Hussain MS, Maqbool M, Arab MM, et al. Transforming hemophilia management: lessons from gene therapy clinical trials. Mol Biotechnol. 2025;68:1597-608.
6. Bhadury R, Athar M, Mishra P, Gogoi C, Sharma S, Ghorpade DS. Development and validation of AAV-mediated liver, liver-VAT, and liver-brain SORT and therapeutic regulation of FASN in hepatic de novo lipogenesis. Cells. 2025;14:372.
7. Zakas PM, Cunningham SC, Doherty A, et al. Sleeping beauty mRNA-LNP enables stable rAAV transgene expression in mouse and NHP hepatocytes and improves vector potency. Mol Ther. 2024;32:3356-71.
8. Herrera-Barrera M, Gautam M, Lokras A, Vlasova K, Foged C, Sahay G. Lipid nanoparticle-enabled intracellular delivery of prime editors. AAPS J. 2023;25:65.
9. Rothgangl T, Tálas A, Ioannidi EI, et al. Treatment of a metabolic liver disease in mice with a transient prime editing approach. Nat Biomed Eng. 2025;9:1705-18.
10. Gracia-Sancho J, Caparrós E, Fernández-Iglesias A, Francés R. Role of liver sinusoidal endothelial cells in liver diseases. Nat Rev Gastroenterol Hepatol. 2021;18:411-31.
11. Wang L, He L, Yi W, et al. ADAMTS18-fibronectin interaction regulates the morphology of liver sinusoidal endothelial cells. iScience. 2024;27:110273.
12. Zhou J, Wang J, Zhang L, Zhang C, Tian C. Defenestration of liver sinusoidal endothelial cells: the trigger of liver fibrosis. Pharmaceuticals. 2025;18:893.
13. Wang N, Guo M, Zhang C, et al. Liver regeneration: unraveling the molecular mechanisms and clinical application. J Transl Med. 2025;23:1409.
14. Gilgenkrantz H, Collin De L'hortet A. Understanding liver regeneration: from mechanisms to regenerative medicine. Am J Pathol. 2018;188:1316-27.
15. Li B, Rodrigo-Torres D, Pelz C, et al. Cell networks in the mouse liver during partial hepatectomy. Stem Cell Rep. 2025;20:102683.
17. Moore HF, Usmani S, Froghi S, et al. Immune cells in liver regeneration: current evidence and potential clinical targets. Int Rev Immunol. 2025;44:353-92.
18. Venzin V, Beccaria CG, Perucchini C, et al. CD4+ T cells license Kupffer cells to reverse CD8+ T cell dysfunction induced by hepatocellular priming. Nat Immunol. 2025;26:1352-66.
19. Ferriero R, Bruno G, Padula A, et al. Impact of liver fibrosis on AAV-mediated gene transfer to mouse hepatocytes. Nat Commun. 2025;16:2118.
20. Acharya P, Chouhan K, Weiskirchen S, Weiskirchen R. Cellular mechanisms of liver fibrosis. Front Pharmacol. 2021;12:671640.
21. Zhou T, Kiran M, Lui KO, Ding Q. Decoding liver fibrogenesis with single-cell technologies. Life Med. 2022;1:333-44.
22. Yang T, Gu Z, Feng J, Shan J, Qian C, Zhuang N. Non-parenchymal cells: key targets for modulating chronic liver diseases. Front Immunol. 2025;16:1576739.
23. Muro AF, D’antiga L, Mingozzi F. Gene therapy in pediatric liver disease. In: D'antiga L, editors. Pediatric hepatology and liver transplantation. Cham: Springer International Publishing; 2019. pp. 799-829.
24. Simini G, Batty P. The hepatic Odyssey of Adeno-associated virus: from gene delivery to long-term outcomes. J Thromb Haemost. 2025;23:3781-90.
25. Dalwadi DA, Calabria A, Tiyaboonchai A, et al. AAV integration in human hepatocytes. Mol Ther. 2021;29:2898-909.
26. Jeyaraj R, Brown LK, Jagadisan B, et al. Safety of in vivo gene therapy in children: mechanisms and management of liver injury. Lancet Child Adolesc Health. 2026;10:448-61.
27. Byrne BJ, Flanigan KM, Matesanz SE, et al. Current clinical applications of AAV-mediated gene therapy. Mol Ther. 2025;33:2479-516.
28. Baruteau J, Cunningham SC, Yilmaz BS, et al. Safety and efficacy of an engineered hepatotropic AAV gene therapy for ornithine transcarbamylase deficiency in cynomolgus monkeys. Mol Ther Methods Clin Dev. 2021;23:135-46.
29. Shi X, Aronson SJ, Ten Bloemendaal L, et al. Efficacy of AAV8-hUGT1A1 with Rapamycin in neonatal, suckling, and juvenile rats to model treatment in pediatric CNs patients. Mol Ther Methods Clin Dev. 2021;20:287-97.
30. Elmore ZC, Oh DK, Simon KE, Fanous MM, Asokan A. Rescuing AAV gene transfer from neutralizing antibodies with an IgG-degrading enzyme. JCI Insight. 2020;5:e139881.
31. Schulz M, Levy DI, Petropoulos CJ, et al. Binding and neutralizing anti-AAV antibodies: detection and implications for rAAV-mediated gene therapy. Mol Ther. 2023;31:616-30.
32. Dhungel BP, Winburn I, Pereira CDF, Huang K, Chhabra A, Rasko JEJ. Understanding AAV vector immunogenicity: from particle to patient. Theranostics. 2024;14:1260-88.
33. Jagadisan B, Dhawan A. Hepatotoxicity in adeno-associated viral vector gene therapy. Curr Hepatol Rep. 2023;22:276-90.
34. Kaiser CW, Rouchka EC, Smith ML. Adaptation of lentiviral vectors for viral gene therapy and their impact on host cell biology. J Transl Med. 2026;24:272.
35. Ranzani M, Cesana D, Bartholomae CC, et al. Lentiviral vector-based insertional mutagenesis identifies genes associated with liver cancer. Nat Methods. 2013;10:155-61.
36. Cesana D, Ranzani M, Volpin M, et al. Uncovering and dissecting the genotoxicity of self-inactivating lentiviral vectors in vivo. Mol Ther. 2014;22:774-85.
37. Baek Y, Seo YR, Kim S, Jung S, Kim CH, Lee Y. From ex vivo to in vivo: advances in lentiviral vector engineering for CAR-T therapy. Immune Netw. 2026;26:e13.
38. Kang DD, Marks A, Morla-Folch J, Dong Y, Brown BD, Teunissen AJP. Targeting and tracking mRNA lipid nanoparticles at the particle, transcript and protein level. Nat Biomed Eng. 2025;9:1591-609.
39. Hosseini-Kharat M, Bremmell KE, Prestidge CA. Why do lipid nanoparticles target the liver? Understanding of biodistribution and liver-specific tropism. Mol Ther Methods Clin Dev. 2025;33:101436.
40. Qiu M, Glass Z, Chen J, et al. Lipid nanoparticle-mediated codelivery of Cas9 mRNA and single-guide RNA achieves liver-specific in vivo genome editing of Angptl3. Proc Natl Acad Sci USA. 2021;118:e2020401118.
41. Zhang T, Yin H, Li Y, et al. Optimized lipid nanoparticles (LNPs) for organ-selective nucleic acids delivery in vivo. iScience. 2024;27:109804.
42. Younis MA, Sato Y, Elewa YH, Kon Y, Harashima H. Self-homing nanocarriers for mRNA delivery to the activated hepatic stellate cells in liver fibrosis. J Controlled Release. 2023;353:685-98.
43. Kim Y, Chatterjee S, Robertson AL, et al. Mannose-conjugated cholesterol containing lipid nanoparticles for active targeted mRNA delivery to liver sinusoidal endothelial and kupffer cells. Bioconjugate Chem. 2025;36:2181-96.
44. Kasiewicz LN, Biswas S, Beach A, et al. GalNAc-lipid nanoparticles enable non-LDLR dependent hepatic delivery of a CRISPR base editing therapy. Nat Commun. 2023;14:2776.
45. Elsharkasy OM, Nordin JZ, Hagey DW, et al. Extracellular vesicles as drug delivery systems: why and how? Adv Drug Delivery Rev. 2020;159:332-43.
46. Serrano DR, Juste F, Anaya BJ, et al. Exosome-based drug delivery: a next-generation platform for cancer, infection, neurological and immunological diseases, gene therapy and regenerative medicine. Pharmaceutics. 2025;17:1336.
47. Sun X, Lian Y, Tian T, Cui Z. Virus-like particle encapsulation of functional proteins: advances and applications. Theranostics. 2024;14:7604-22.
48. Zhang J, Yang X, Chang Z, Zhu W, Ma Y, He H. Polymeric nanocarriers for therapeutic gene delivery. Asian J Pharm Sci. 2025;20:101015.
49. Cavazza A, Molina-Estévez FJ, Reyes ÁP, et al. Advanced delivery systems for gene editing: a comprehensive review from the GenE-HumDi COST Action Working Group. Mol Ther Nucl Acids. 2025;36:102457.
50. Canepari C, Milani M, Simoni C, et al. Enhancing the potency of in vivo lentiviral vector mediated gene therapy to hepatocytes. Nat Commun. 2025;16:4802.
51. Simoni C, Nozi J, Starinieri F, et al. Liver fibrosis negatively impacts in vivo gene transfer to murine hepatocytes. Nat Commun. 2025;16:2119.
52. Wang J, Gessler DJ, Zhan W, Gallagher TL, Gao G. Adeno-associated virus as a delivery vector for gene therapy of human diseases. Sig Transduct Target Ther. 2024;9:78.
53. D’antiga L, Beuers U, Ronzitti G, et al. Gene therapy in patients with the crigler-najjar syndrome. N Engl J Med. 2023;389:620-31.
54. Schambach A, Buchholz CJ, Torres-Ruiz R, et al. A new age of precision gene therapy. Lancet. 2024;403:568-82.
55. Ay C, Reinisch A. Gene therapy: principles, challenges and use in clinical practice. Wien Klin Wochenschr. 2024;137:261-71.
56. Sayed N, Allawadhi P, Khurana A, et al. Gene therapy: comprehensive overview and therapeutic applications. Life Sci. 2022;294:120375.
57. Kosicki M, Tomberg K, Bradley A. Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat Biotechnol. 2018;36:765-71.
58. Haapaniemi E, Botla S, Persson J, Schmierer B, Taipale J. CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat Med. 2018;24:927-30.
59. Xu W, Zhang S, Qin H, Yao K. From bench to bedside: cutting-edge applications of base editing and prime editing in precision medicine. J Transl Med. 2024;22:1133.
60. Murray JB, Harrison PT, Scholefield J. Prime editing: therapeutic advances and mechanistic insights. Gene Ther. 2024;32:83-92.
61. Simoni C, Barbon E, Muro AF, Cantore A. In vivo liver targeted genome editing as therapeutic approach: progresses and challenges. Front Genome Ed. 2024;6:1458037.
62. Whittaker MN, Testa LC, Quigley A, et al. Improved specificity and efficiency of in vivo adenine base editing therapies with hybrid guide RNAs. Nat Biomed Eng. 2025:1545.
63. Khirallah J, Bloomer H, Wich D, et al. In vivo base editing of Angptl3 via lipid nanoparticles to treat cardiovascular disease. Mol Ther Nucl Acids. 2025;36:102486.
64. Gillmore JD, Gane E, Taubel J, et al. CRISPR-Cas9 in vivo gene editing for transthyretin amyloidosis. N Engl J Med. 2021;385:493-502.
65. Liu D, Cao D, Han R. Recent advances in therapeutic gene-editing technologies. Mol Ther. 2025;33:2619-44.
66. Xiong H, Guo J. Targeting hepatic stellate cells for the prevention and treatment of liver cirrhosis and hepatocellular carcinoma: strategies and clinical translation. Pharmaceuticals. 2025;18:507.
67. Martínez-García J, Molina A, González-Aseguinolaza G, Weber ND, Smerdou C. Gene therapy for acquired and genetic cholestasis. Biomedicines. 2022;10:1238.
68. Yang AY, Wistuba-Hamprecht K, Greten TF, Ruf B. Innate-like T cells in liver disease. Trends Immunol. 2024;45:535-48.
69. De Sabbata G, Boisgerault F, Guarnaccia C, et al. Long-term correction of ornithine transcarbamylase deficiency in Spf-Ash mice with a translationally optimized AAV vector. Mol Ther Methods Clin Dev. 2021;20:169-80.
70. Seker Yilmaz B, Gissen P. Genetic therapy approaches for ornithine transcarbamylase deficiency. Biomedicines. 2023;11:2227.
71. Weiss JN. Ornithine transcarbamylase deficiency. In: Neurologic gene therapy. Cham: Springer Nature Switzerland; 2025. pp. 421-9.
72. Sambati V, Laudisio S, Motta M, Esposito S. Therapeutic options for crigler-najjar syndrome: a scoping review. Int J Mol Sci. 2024;25:11006.
73. Kishnani PS, Sun B, Koeberl DD. Gene therapy for glycogen storage diseases. Hum Mol Genet. 2019;28:R31-41.
74. Chou JY, Mansfield BC. Gene therapy and genome editing for type I glycogen storage diseases. Front Mol Med. 2023;3:1167091.
75. Moreno D, Murillo O, Gazquez C, et al. Visualization of the therapeutic efficacy of a gene correction approach in Wilson’s disease by laser-ablation inductively coupled mass spectrometry. J Hepatol. 2018;68:1088-90.
76. Zeng C, Lin Y, Lu X, et al. Evaluation of efficacy and safety of AAV8-ΔC4ATP7B gene therapy in a mutant mouse model of Wilson’s disease. Mol Ther Methods Clin Dev. 2025;33:101435.
77. Bosma PJ, Wits M, Oude-Elferink RPJ. Gene therapy for progressive familial intrahepatic cholestasis: current progress and future prospects. Int J Mol Sci. 2020;22:273.
78. Weber ND, Odriozola L, Martínez-García J, et al. Gene therapy for progressive familial intrahepatic cholestasis type 3 in a clinically relevant mouse model. Nat Commun. 2019;10:5694.
79. Ermi AG, Younis RM, Rodriguez K, Sarkar D. Gene therapy strategies for hepatocellular carcinoma (HCC): current landscape and future directions. Cancers. 2025;17:3608.
80. Zhang J, Xiao Y, Zhang J, Yang Y, Zhang L, Liang F. Recent advances of engineered oncolytic viruses-based combination therapy for liver cancer. J Transl Med. 2024;22:3.
81. Shen Y, Bai X, Zhang Q, et al. Oncolytic virus VG161 in refractory hepatocellular carcinoma. Nature. 2025;641:E5.
82. Zhou Y, Wei S, Xu M, et al. CAR-T cell therapy for hepatocellular carcinoma: current trends and challenges. Front Immunol. 2024;15:1489649.
83. Hong J, Yu J, Buratto D, et al. Unveiling the role of mechanical microenvironment in hepatocellular carcinoma: molecular mechanisms and implications for therapeutic strategies. Int J Biol Sci. 2024;20:5239-53.
84. Nathwani AC, Tuddenham EG, Rangarajan S, et al. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N Engl J Med. 2011;365:2357-65.
85. George LA, Sullivan SK, Giermasz A, et al. Hemophilia B gene therapy with a high-specific-activity factor IX variant. N Engl J Med. 2017;377:2215-27.
86. Pasi KJ, Rangarajan S, Mitchell N, et al. Multiyear follow-up of AAV5-hFVIII-SQ gene therapy for hemophilia A. N Engl J Med. 2020;382:29-40.
87. Lim J, Choi SJ, Gao F, Kishnani PS, Sun B. A novel gene therapy approach for GSD III using an AAV vector encoding a bacterial glycogen debranching enzyme. Mol Ther Methods Clin Dev. 2020;18:240-9.
88. Chandler RJ, Di Pasquale G, Sloan JL, et al. Systemic gene therapy for methylmalonic acidemia using the novel adeno-associated viral vector 44.9. Mol Ther Methods Clin Dev. 2022;27:61-72.
89. Chandler RJ, Di Pasquale G, Choi E, et al. Systemic gene therapy using an AAV44.9 vector rescues a neonatal lethal mouse model of propionic acidemia. Mol Ther Methods Clin Dev. 2023;30:181-90.
90. Brantly ML, Chulay JD, Wang L, et al. Sustained transgene expression despite T lymphocyte responses in a clinical trial of rAAV1-AAT gene therapy. Proc Natl Acad Sci USA. 2009;106:16363-8.
91. Yao ZQ, Schank MB, Zhao J, et al. The potential of HBV cure: an overview of CRISPR-mediated HBV gene disruption. Front Genome Ed. 2024;6:1467449.
92. Hsueh Y, Chang Y, Loh C, Gershwin ME, Chuang Y. AAV-IL-22 modifies liver chemokine activity and ameliorates portal inflammation in murine autoimmune cholangitis. J Autoimmun. 2016;66:89-97.
93. Zhang VX, Tsui Y, Ng IO. MASLD/MASH—New mechanisms and treatments. Hepatol Commun. 2025;9:e0770.
94. Liu S, Ji Y, Wei L, et al. Crotonylation of IDH1 alleviates MASLD progression by enhancing the TCA cycle. Nat Commun. 2025;16:7961.
95. Gross D, Tedesco N, Leborgne C, Ronzitti G. Overcoming the challenges imposed by humoral immunity to AAV vectors to achieve safe and efficient gene transfer in seropositive patients. Front Immunol. 2022;13:857276.
96. Bakos T, Mészáros T, Kozma GT, et al. mRNA-LNP COVID-19 vaccine lipids induce complement activation and production of proinflammatory cytokines: mechanisms, effects of complement inhibitors, and relevance to adverse reactions. Int J Mol Sci. 2024;25:3595.
97. Saxena S, Sharma S, Kumar G, Thakur S. Unravelling the complexity of CARPA: a review of emerging advancements in therapeutic strategies. Arch Dermatol Res. 2025;317:439.
98. Assaf BT. Systemic toxicity of recombinant adeno-associated virus gene therapy vectors. Toxicol Pathol. 2024;52:523-30.
100. Anzalone AV, Randolph PB, Davis JR, et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576:149-57.
101. Reiss UM, Davidoff AM, Tuddenham EG, et al. Sustained clinical benefit of AAV gene therapy in severe hemophilia B. N Engl J Med. 2025;392:2226-34.
102. Clément N, Grieger JC. Manufacturing of recombinant adeno-associated viral vectors for clinical trials. Mol Ther Methods Clin Dev. 2016;3:16002.
103. Hou X, Zaks T, Langer R, Dong Y. Lipid nanoparticles for mRNA delivery. Nat Rev Mater. 2021;6:1078-94.
104. Wec AZ, Lin KS, Kwasnieski JC, Sinai S, Gerold J, Kelsic ED. Overcoming immunological challenges limiting capsid-mediated gene therapy with machine learning. Front Immunol. 2021;12:674021.
105. Segers S. Heritable genome editing: ethical aspects of a developing domain. Hum Reprod. 2023;38:2055-61.
106. Iyer AA, Saade D, Bharucha-Goebel D, et al. Ethical challenges for a new generation of early-phase pediatric gene therapy trials. Genet Med. 2021;23:2057-66.
107. Yang W, Wang A, Liu B. Adeno-associated virus gene therapy for hemophilia: an update meta-analysis and systematic review. Front Med. 2025;12:1580264.
108. Barnes C, Scheideler O, Schaffer D. Engineering the AAV capsid to evade immune responses. Curr Opin Biotechnol. 2019;60:99-103.
109. Eid F, Chen AT, Chan KY, et al. Systematic multi-trait AAV capsid engineering for efficient gene delivery. Nat Commun. 2024;15:6602.
110. Gao H, Gao S, Kan G, Valentovich LN, An Y. Research progress of base editing and prime editing tools based on the CRISPR/Cas system. Mol Ther Nucleic Acids. 2025;36:102771.
111. Booth BJ, Nourreddine S, Katrekar D, et al. RNA editing: expanding the potential of RNA therapeutics. Mol Ther. 2023;31:1533-49.
112. Jin G, Lim B. Epigenome editing and epigenetic gene regulation in disease phenotypes. Korean J Chem Eng. 2022;39:1361-7.
113. Rezvani M, Español-Suñer R, Malato Y, et al. In vivo hepatic reprogramming of myofibroblasts with AAV vectors as a therapeutic strategy for liver fibrosis. Cell Stem Cell. 2016;18:809-16.
114. Jalan-Sakrikar N, Brevini T, Huebert RC, Sampaziotis F. Organoids and regenerative hepatology. Hepatology. 2023;77:305-22.
115. Adams MT, Jasper H, Mosteiro L. Unlocking regeneration: how partial reprogramming resembles tissue healing. Curr Opin Genet Dev. 2025;93:102351.
116. Hashem EM, Karrar AM, Mabrouk MS. Advancing liver cancer diagnosis and treatment with multi-omics approaches: a systematic review. Discov Onc. 2025;16:2188.
117. Lu X, Li Y, Li Y, et al. Advances of multi-omics applications in hepatic precancerous lesions and hepatocellular carcinoma: the role of extracellular vesicles. Front Mol Biosci. 2023;10:1114594.
118. Rao J, Li Y, Leng K. Artificial intelligence in hepatocellular carcinoma screening: applications and challenges. Front Med. 2026;12:1713887.
119. Malik S, Das R, Thongtan T, Thompson K, Dbouk N. AI in hepatology: revolutionizing the diagnosis and management of liver disease. J Clin Med. 2024;13:7833.
120. Joseph AM, Karas M, Ramadan Y, Joubran E, Jacobs RJ. Ethical perspectives of therapeutic human genome editing from multiple and diverse viewpoints: a scoping review. Cureus. 2022;14:e31927.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].