



Artificial intelligence and circulating tumor DNA minimal residual disease in perioperative resectable non-small cell lung cancer
 0
0 Abstract
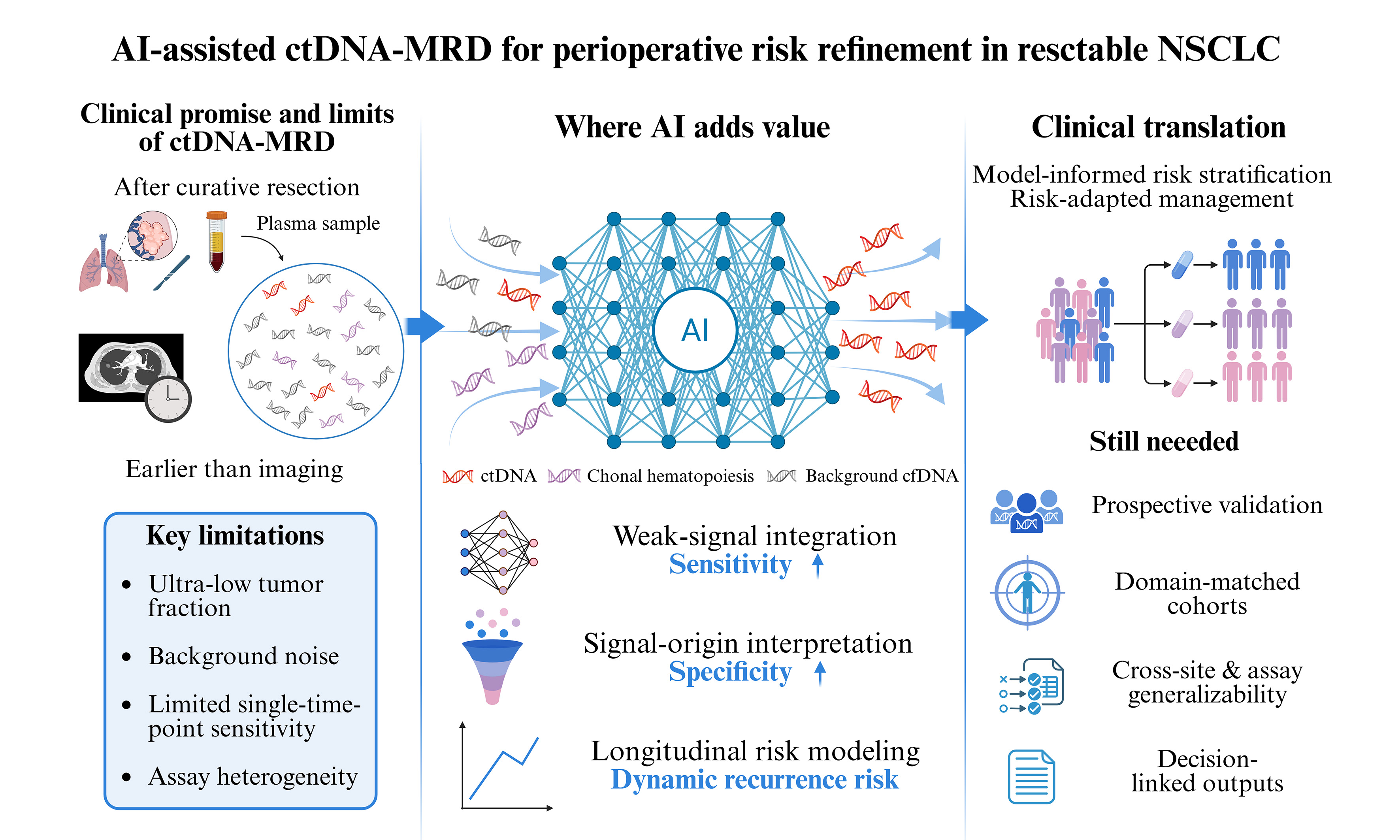
Despite curative-intent treatment, recurrence risk in resectable non-small cell lung cancer (NSCLC) remains difficult to define, and postoperative management still depends on imperfect clinicopathologic assessment. Circulating tumor DNA (ctDNA)-based minimal residual disease (MRD) has emerged as a promising adjunct for postoperative risk stratification, early relapse detection, and perioperative response assessment. Its clinical translation, however, is still limited by low sensitivity in low-shedding diseases, imperfect specificity from biological background signals, interpretive uncertainty, and a lack of assay standardization. In this context, artificial intelligence (AI) is relevant not as a generic add-on, but as a potential means to improve weak-signal detection, refine variant-origin assignment, and translate serial ctDNA measurements into clinically interpretable risk estimates. This review considers the current clinical role of ctDNA-MRD in perioperative NSCLC, the barriers to routine implementation, and the extent to which AI may help make molecular monitoring more reliable and clinically useful.
Keywords
INTRODUCTION
Lung cancer remains the leading cause of cancer death worldwide, with nearly 2.5 million new cases and 1.8 million deaths estimated in 2022[1]. Even among patients with resectable non-small cell lung cancer (NSCLC) treated with curative intent, durable disease control remains limited: postoperative recurrence has long been reported in 30%-55% of cases, and a recent meta-analysis still showed 5-year recurrence-free survival declining from 82% in stage I disease to 34% in stage III[2,3]. This persistent gap between apparently curative treatment and residual biologic risk has driven interest in molecular approaches that can refine postoperative risk assessment beyond stage, pathology, and imaging alone.
Accordingly, circulating tumor DNA (ctDNA)-based minimal residual disease (MRD) has attracted growing interest as a non-invasive tool for postoperative risk assessment, recurrence surveillance, and perioperative decision support. However, it is not yet sufficiently standardized or validated to serve as a stand-alone biomarker for direct clinical decision-making. Its utility remains limited by weak postoperative tumor signal, background noise, variable result interpretation, and the lack of standardized analytical and clinical frameworks. The integration of artificial intelligence (AI) with ctDNA analysis may further expand this potential by improving signal detection and supporting more refined interpretation and risk estimation. This review examines the clinical value and current limitations of ctDNA-MRD in perioperative NSCLC, and discusses how AI may help address these gaps, where it still falls short, and what will be required for clinical translation.
Literature search strategy for this narrative review
For this narrative review, we searched PubMed, Embase, Web of Science, and ClinicalTrials.gov for English-language literature published between January 2015 and October 2025. Keywords included “non-small cell lung cancer”, “perioperative”, “resectable”, “circulating tumor DNA”, “minimal residual disease”, “artificial intelligence”, and “machine learning”.
CURRENT ROLE OF PERIOPERATIVE ctDNA-MRD
Clinical rationale
MRD generally refers to tumor cells that remain in the body after curative-intent treatment but are still below the detection limit of conventional methods[4-7]. ctDNA is the tumor-derived fraction of circulating cell-free DNA (cfDNA) and carries molecular features of the primary tumor[8]. It therefore provides a practical liquid-biopsy readout of residual molecular disease. In perioperative NSCLC, ctDNA-detected MRD is therefore best interpreted as a molecular surrogate of residual tumor burden after curative-intent treatment.
In resectable NSCLC, ctDNA status at the postoperative landmark time point is significantly associated with an increased risk of recurrence and death. This landmark assessment is usually performed between 2 weeks and 4 months after surgery. Gale et al. reported that patients with detectable ctDNA at this time point had a 14.8-fold higher risk of recurrence and a 5.48-fold higher risk of death[9].
The importance of ctDNA-MRD in the perioperative setting also lies in its temporal advantage over imaging. During longitudinal follow-up, ctDNA monitoring identified 79% of patients who eventually relapsed before or at the time of radiographic progression, whereas the detection rate of a single postoperative test was only 41%[10]. In the TRACERx study, ctDNA signaled relapse in early-stage NSCLC a median of 119 days earlier than computed tomography (CT) imaging[11]. An exploratory analysis from ADAURA likewise showed that MRD positivity preceded imaging-defined disease-free survival events by a median of 4.7 months[12]. Against this background, perioperative ctDNA-MRD is best understood as a risk stratification tool. It refines recurrence-risk assessment, helps distinguish biologically high-risk from low-risk postoperative states, and supports more individualized follow-up and treatment discussions.
Current assessment strategies
To date, a range of MRD detection assays have been developed, and these methods differ in sensitivity, specificity, turnaround time, susceptibility to technical bias, and the degree of laboratory standardization [Table 1][13-18]. In practice, ctDNA-MRD detection mainly follows two broad strategies: tumor-informed and tumor-naïve, distinguished by whether prior tumor sequencing is required. Current technical approaches are mainly based on droplet digital polymerase chain reaction (ddPCR) or deep next-generation sequencing (NGS), often combined with unique molecular identifier (UMI)-based designs and other error-suppression strategies to improve interpretive reliability when ctDNA approaches the limit of detection[19].
Comparison of ctDNA-MRD assay strategies and analytical methods
| Category | Approach | Principle/what it captures | Main strengths | Main limitations or trade-offs | Representative reference |
| Assay strategy | TI | Uses patient-specific tumor variants identified from tumor tissue and then tracks those variants in plasma | Highest specificity; better suited to ultra-low-burden MRD settings | Requires tumor tissue; longer turnaround; higher cost; less flexible for emergent variants | Abbosh et al., 2017[13] |
| Assay strategy | TN | Uses predefined plasma-based genomic or epigenomic features without prior tumor sequencing | Faster workflow; no tissue dependence; easier to scale | Lower individualization may reduce sensitivity; more background noise; interpretation is harder in low-shedding disease | Hong et al., 2024[14] |
| Analytical approach | ddPCR | Allele-specific PCR in microdroplets for highly focused detection of known mutations | Very high analytical sensitivity; absolute quantification; low LoD for predefined loci | Narrow genomic scope; limited to known targets; less suited to heterogeneous residual disease | Gassa et al., 2021[15] |
| Analytical approach | NGS-based assays | Deep sequencing of multiple loci or genomic regions, often with hybrid-capture or amplicon-based designs | Broader genomic coverage; enables multi-mutation tracking and longitudinal monitoring | Requires deeper sequencing, stronger error suppression, and more complex bioinformatics | Chaudhuri et al., 2017[16] |
| Analytical refinement | UMI-enabled/bioinformatic error suppression | Molecular barcoding plus computational filtering to suppress background artifacts and improve low-VAF calling | Improves analytical specificity and confidence at very low allele fractions | Still does not fully eliminate false negatives; performance depends on assay design and cfDNA input | Newman et al., 2016[17] |
| Analytical refinement | Phased-variant ultra-sensitive methods | Tracks multiple mutations on the same DNA fragment to lower background error far beyond single-SNV methods | Extremely high analytical sensitivity in ultra-low-signal settings | Technically demanding; real-world perioperative NSCLC validation is still limited | Kurtz et al., 2021[18] |
Current clinical applications
Postoperative risk stratification
ctDNA positivity after resection in NSCLC is clinically meaningful because recurrence is usually both more likely and earlier once molecular disease becomes detectable. This temporal lead over imaging can be clinically meaningful. In the cohort reported by Peng et al., 19 of 30 postoperative ctDNA-positive patients (63.3%) later developed clinical recurrence, and ctDNA preceded radiographic findings or symptoms by a median of 12.6 months[20]. Qiu et al. reported postoperative ctDNA positivity in 18 of 85 patients (21.2%); during surveillance, ctDNA became detectable in 27 of 34 patients who eventually relapsed and preceded radiographic recurrence by a median of 88 days[10].
A single postoperative landmark test also has clear limitations. It can separate higher- and lower-risk groups, but it does not fully support individualized decision-making, particularly in low-shedding or slower-growing tumors, where current assays still miss a proportion of biologically relevant disease[21]. In the prospective cohort reported by Zhang et al., landmark MRD yielded a negative predictive value (NPV) of 86.6% and a positive predictive value (PPV) of 81.0%. Once serial time points were incorporated, these increased to 96.8% and 89.1%, respectively[22]. These data provide a stronger rationale for longitudinal monitoring than a general appeal to “dynamic assessment” alone.
Adjuvant treatment stratification
For adjuvant treatment, ctDNA is better understood as a marker that enriches for biologically high-risk disease than as a stand-alone treatment switch. Available evidence supports this risk-enrichment role. In LUNGCA-1, MRD-positive patients who received adjuvant therapy had better recurrence-free survival than MRD-positive patients who did not, and after adjustment this association remained significant in the MRD-positive subgroup[23]. Chen et al. reported a similar pattern: among patients with positive ctDNA at an early postsurgical time point (P2), adjuvant therapy was associated with a median RFS of 269 days, compared with 111 days in those who did not receive it[24]. Qiu et al. likewise showed that, in stage II-III disease, the postsurgical ctDNA-positive group benefited from adjuvant chemotherapy (ACT), whereas ctDNA-negative patients had a low risk of relapse regardless of whether ACT was administered[10].
Nevertheless, ctDNA status should not yet be used alone to define escalation or de-escalation strategies. In TRACERx, postoperative ctDNA profiling appeared to capture not only residual disease but, in some cases, resistance to adjuvant chemotherapy, with ctDNA burden continuing to rise despite treatment[11]. Exploratory analyses from IMpower010 add the same note of caution. During the first two years, about one-third of recurrences occurred in patients who were ctDNA-negative after surgery and chemotherapy, and longer-term analyses suggested benefit from adjuvant atezolizumab regardless of ctDNA status[25]. Thus, ctDNA positivity helps identify patients at higher biological risk, whereas ctDNA negativity remains insufficient to determine who can safely forgo systemic therapy.
Perioperative response assessment
In the neoadjuvant and perioperative setting, changes in ctDNA status over time may be more informative than a single binary result. ctDNA clearance illustrates this principle most clearly. In NADIM, clearance after neoadjuvant nivolumab plus chemotherapy was associated with markedly longer progression-free survival (PFS) and overall survival (OS), with hazard ratios (HR) of 0.16 and 0.05, respectively[26]. A similar pattern was seen in AEGEAN: after three cycles of perioperative durvalumab plus chemotherapy, ctDNA clearance identified patients with better event-free survival, particularly in the durvalumab arm, with an HR of 0.26[27,28]. This distinction is clinically relevant. A postoperative snapshot indicates whether molecular disease is detectable at a single time point, whereas ctDNA clearance captures whether the tumor-derived signal is decreasing during treatment. Nevertheless, postoperative ctDNA status itself may also carry important prognostic value in the perioperative setting. In a prospective cohort of patients receiving neoadjuvant immunotherapy-based regimens including chemoimmunotherapy, postoperative ctDNA positivity was independently associated with recurrence (HR 5.37, 95%CI: 1.27-22.67; P = 0.01)[29]. These findings indicate that postoperative ctDNA status may complement ctDNA clearance in identifying patients at increased risk of relapse following neoadjuvant therapy. Together, these biomarkers provide a more comprehensive assessment of treatment response and recurrence risk than either metric alone.
Overall, ctDNA-based MRD provides a molecular framework for refining adjuvant and consolidation therapy strategies. However, prospective interventional trials remain essential for establishing standardized MRD-guided treatment algorithms. Numerous ongoing studies are exploring MRD-based interventions. Summaries of 20 completed studies and 42 ongoing trials are provided in Supplementary Tables 1 and 2. Additionally, Supplementary Tables 3 and 4 respectively summarize trials that utilize AI/machine learning (ML) for ctDNA analysis in NSCLC and trials that apply AI/ML to liquid biopsy analysis in NSCLC.
LIMITS OF CONVENTIONAL ctDNA-MRD
The principal limitation of ctDNA-based MRD in resectable NSCLC remains sensitivity. After curative-intent treatment, the tumor-derived fraction can fall to ≤ 0.01%-0.1% of total cfDNA, therefore requiring assays to detect an intrinsically scarce biological signal[30]. This biological constraint helps explain the wide variability in reported postoperative detection rates. In the 13-study systematic review cited by Boukouris, reported MRD detection rates ranged from 6% to 46%[31]. Longitudinal monitoring improves performance, but it does not remove the limitation. In the meta-analysis by Zhong et al., pooled sensitivity for longitudinal surveillance was 76% for hybrid-capture NGS and 77% for amplicon-based NGS[32]. This limitation is particularly evident in low-shedding disease. Zhang et al. showed that brain-only recurrence was detected in only 1 of 5 cases during longitudinal monitoring[22]. Chaudhuri et al. showed that broader variant tracking can improve detection: when cancer personalized profiling by deep sequencing (CAPP-seq) tracked all known variants rather than a single mutation, MRD detection increased from 58% to 94%[16]. Even this finding is better interpreted as evidence that sensitivity can be improved, rather than as proof that current assays are sufficient across postoperative NSCLC.
Specificity also remains imperfect. Plasma does not contain tumor DNA alone, and clonal hematopoiesis (CH) is the clearest competing source of low-frequency variants. This matters even more in MRD analysis, because the variant allele frequency (VAF) range being interrogated is already close to the technical floor[33]. In the OAK correlative analysis, clonal hematopoiesis of indeterminate potential (CHIP)-derived variants were detectable in paired plasma and peripheral blood mononuclear cells (PBMC) samples from patients with late-stage NSCLC; the number of CHIP variants increased with age, and tumor protein p53 (TP53) was the most frequently mutated gene[34]. Thus, a plasma mutation may be analytically real but biologically unrelated to residual tumor. Without matched leukocyte sequencing or explicit CHIP filtering, this creates a clear interpretive risk: a positive plasma call may reflect hematopoietic background rather than persistent NSCLC[35]. Tumor-naïve and plasma-only workflows are especially exposed here, because the detected variants are not directly anchored to the resected tumor[36]. The opposite problem also remains: a negative plasma result may simply reflect limited ctDNA shedding rather than the absence of residual disease.
Methodological heterogeneity represents another major barrier. Studies grouped under the term “ctDNA-MRD” in NSCLC still differ substantially in platform, threshold, postoperative sampling window, and patient population. Boukouris specifically noted that performance varies with disease stage, molecular background, treatment exposure, follow-up duration, assay type, and sampling time after surgery. The same review also makes clear that tumor-informed and tumor-naïve approaches are not interchangeable choices[31]. Tumor-informed assays generally gain specificity by anchoring plasma findings to known tumor variants, whereas tumor-naïve assays may be faster and easier to deploy but accept more biological and analytical background noise[11,13,37-39]. A practical unresolved issue is that the field still lacks consensus on two basic questions: when postoperative blood should be drawn for clinically meaningful MRD assessment, and what ctDNA level should count as residual disease. Until those points are standardized, study results will continue to look stronger in some cohorts than in others, and thresholds from one series will remain difficult to transfer into routine care.
Another major limitation lies in clinical actionability. Although prognostic value has been shown repeatedly, decision-grade utility requires a higher evidentiary standard that has not yet been met in NSCLC. Boukouris notes that ctDNA-MRD has still not been incorporated into official guidelines for adaptive decision-making in early-stage disease[31]. Part of the reason is the persistent weakness of a negative result. ctDNA clearance is informative, but it is not the same as confirmed eradication of residual disease. In exploratory biomarker analyses from CheckMate 816 and CheckMate 77T, ctDNA clearance was associated with higher pathologic complete response (pCR) rates; however, pCR still occurred in only 46% of ctDNA-cleared patients in CheckMate 816 and 50% of ctDNA-cleared patients in the nivolumab arm of CheckMate 77T[40-42]. At present, ctDNA is best viewed as a tool for refining postoperative risk stratification and enriching for biologically higher-risk groups. It is not yet strong enough on its own to justify treatment withholding or to serve as a stand-alone trigger for escalation or de-escalation.
WHERE AI ADDS VALUE
Enhancing sensitivity through weak-signal integration
In resectable NSCLC, postoperative ctDNA-based MRD testing is performed in a setting in which the tumor-derived signal is often extremely scarce. After curative-intent treatment, tumor-derived DNA may fall near the lower limit of assay resolution, where conventional platforms become less stable. In the SEQC2 analytical validity study, variants above approximately 0.5% VAF were detected with high sensitivity and reproducibility across assays, whereas performance below this level became less reliable and more variable, particularly when input material was limited[43]. This analytical constraint provides the technical rationale for AI-based ctDNA approaches. The key issue is not merely that conventional assays have limited sensitivity, but that threshold-based, locus-by-locus interpretation uses only part of the available cfDNA information. When ctDNA is sparse, weak but potentially informative signals may still be distributed across fragmentomic patterns, methylation states, and broader genomic features, even when no single locus is confidently positive[30]. AI-based models aim to recover such information by integrating evidence across many reads and feature classes at the sample level, rather than relying solely on whether a predefined mutation exceeds a fixed cutoff[44].
Bahado-Singh et al. provided an early example of this feature-integration strategy in a cfDNA methylation study for lung cancer detection[45]. Using genome-wide methylation profiling with the Illumina EPIC array, they selected informative cytosine-phosphate-guanine (CpG) markers and trained several supervised learning algorithms on multivariable methylation feature sets rather than on individually thresholded loci. The cohort was small, including only 10 lung cancer cases and 20 controls, so these results require cautious interpretation. In repeated resampling analyses, several classifiers achieved area under the curves (AUCs) of 1.00, and the deep-learning model achieved 100% sensitivity and 100% specificity. These results should be interpreted cautiously, because the cohort included only 10 lung cancer cases and 20 controls, and such perfect performance in a small dataset may reflect overfitting rather than clinical maturity. Nevertheless, the study is useful as a proof of principle: when plasma cfDNA is represented as a coordinated methylation pattern, tumor-associated signal may become easier to detect than under a marker-by-marker framework[45].
Kim et al. extended this concept by integrating methylation and fragment-size information in lung cancer plasma[46]. Using a targeted enzymatic methylation sequencing (EM-seq) panel, they generated data from 142 patients with NSCLC and 56 healthy controls. For each sample, they constructed a two-dimensional methylation-fragment size representation and used it as input for a convolutional neural network, enabling methylation and fragment-length information to be interpreted jointly rather than as separate feature classes. The resulting model achieved an AUC of 0.87 and an accuracy of 81.5%, outperforming models based on methylation or fragment-length features alone. The dilution experiment is particularly relevant to the MRD setting: detection was achievable at a tumor fraction of 1% with 98% specificity and at 0.1% with 80% specificity. Although this framework does not by itself solve postoperative MRD detection, it supports the premise that joint modeling can extract weak cfDNA signals that may be difficult to recover when methylation, fragment size, and variant calls are analyzed separately[46].
This direction is further illustrated by MRD-focused platforms such as MRD-EDGE. Widman et al. used plasma whole-genome sequencing (WGS) with machine-learning-guided signal enrichment to integrate single-nucleotide variant (SNV)-level and copy-number variant (CNV)-level information, rather than relying on a single feature type[47]. In analytical testing, MRD-EDGE improved SNV signal enrichment by approximately 300-fold compared with the earlier MRDetect approach and reduced the degree of aneuploidy required for ultrasensitive CNV detection from 1 Gb to 200 Mb[47,48]. In stage III colorectal cancer, preoperative detection reached an AUC of 0.998. More relevant to NSCLC, the same platform was applied to 22 patients with early-stage NSCLC treated with neoadjuvant durvalumab with or without stereotactic body radiation therapy (SBRT), followed by surgery. Pretreatment ctDNA detection achieved an AUC of 0.98, with ctDNA undetectable in only two patients with clinical stage IA disease. Among 14 resected patients included in the survival analysis, recurrence occurred in 5 of 8 MRD-positive patients, whereas none of the 6 MRD-negative patients relapsed during the available follow-up. Although these numbers are small, the findings suggest that whole-genome signal integration may expand the postoperative detection window in settings where conventional high-sensitivity assays remain limited[47].
The most directly relevant evidence comes from postoperative NSCLC cohorts. Wang et al. applied WGS-based cfDNA fragmentomics to 87 patients with resected NSCLC, none of whom had received neoadjuvant therapy, and modeled fragment-size-ratio features at landmark approximately 7-day and 6-month time points[49]. At a specificity of at least 90%, the fragmentomic models achieved sensitivities of 43.5% and 57.9% at the two respective time points, compared with 26.1% and 31.6% for a tumor-informed mutation-based assay applied to the same samples. Combining both approaches across both time points increased overall sensitivity to 78.3%, and high-risk prediction preceded radiographic recurrence by a median of 293 days. Because this study directly examined a postoperative landmark MRD setting in NSCLC, it provides one of the closest analogs to the perioperative task. It suggests that genome-wide fragment integration can recover residual-disease signals missed by locus-specific mutation tracking. However, the cohort was small, and the model was evaluated using leave-one-out cross-validation without independent external validation; therefore, these results should be considered proof-of-principle evidence rather than a definitive estimate of clinical performance[49]. Tumor-naïve fragment-length modeling provides a complementary line of evidence. Zhu et al. developed Fragle, a deep-learning model that estimates ctDNA levels from the density distribution of cfDNA fragment lengths using low-pass WGS data[50]. In 162 patients with resected lung cancer sampled at the landmark time point, Fragle inferred ctDNA levels without requiring tumor tissue. Among 158 patients who were classified as ctDNA-negative by a tumor-agnostic mutation panel, Fragle identified a subgroup with higher inferred ctDNA levels and significantly worse disease-free survival. These findings suggest that fragment-length patterns may provide postoperative risk information even when mutation-based plasma testing is negative. However, this analysis should be interpreted as supportive rather than definitive MRD evidence, because it represents a downstream use of a ctDNA quantification model, involved few recurrence events, and did not test CH discrimination in high-CH populations[50].
The three principal ways in which AI/ML may enhance ctDNA-based MRD assessment in perioperative NSCLC are summarized in Figure 1.
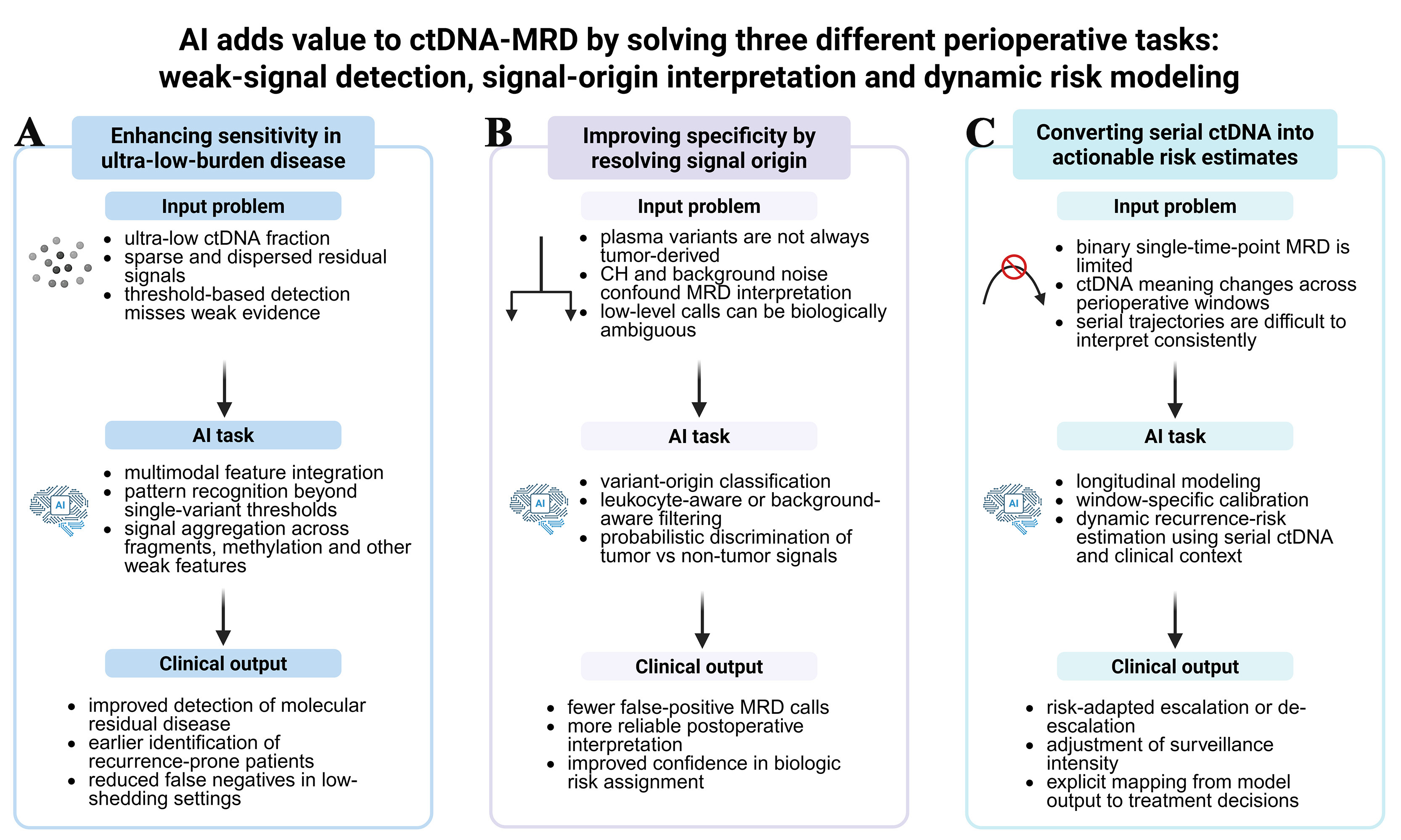
Figure 1. Three principal ways in which AI/ML can improve ctDNA-based MRD assessment in perioperative NSCLC. (A) illustrates the use of AI for weak signal integration to improve sensitivity in ultra-low-burden disease; (B) shows how AI can improve specificity by distinguishing tumor-derived signals from clonal hematopoiesis and other background noise; (C) summarizes longitudinal and window-aware modeling strategies that translate serial ctDNA measurements into dynamic recurrence-risk estimates and clinically actionable management categories. Created in BioRender. Chen, Y. (2026) https://BioRender.com/it4i2ml. NSCLC: Non-small cell lung cancer; ctDNA: circulating tumor DNA; MRD: minimal residual disease; AI: artificial intelligence; ML: machine learning; CH: clonal hematopoiesis.
Improving specificity through signal-origin interpretation
Loss of specificity in ctDNA-based MRD does not arise solely from technical assay limitations. It also fails because plasma contains DNA from multiple biological sources, and CH is one of the most important of them. CH arises from age-associated somatic mutations in hematopoietic stem and progenitor cells, followed by clonal expansion, and many of these variants are easily detectable in plasma cfDNA. This creates a direct challenge for MRD interpretation: VAF alone often does not separate CH-derived variants from true tumor-derived ctDNA, because the two distributions overlap. Consequently, CH can produce false molecular positivity even when sequencing is analytically robust. Razavi et al. made this point clearly in pan-cancer cfDNA profiling, showing that a large fraction of plasma variants had features consistent with CH rather than tumor origin[51]. In NSCLC, this concern has been demonstrated empirically. In the OAK cohort,
That is also why several groups have started to treat variant-origin assignment as a classification problem rather than a manual filtering step. Fairchild et al. pushed this further in a large real-world oncology cohort[36]. Using labeled cfDNA variants and matched sequencing data in a training subset, they developed a machine-learning framework to distinguish CH from tumor-derived alterations and then applied it across 4,324 clinical cfDNA samples. About 30.3% of patients had evidence of CH in plasma, and the prevalence rose with age. Clinically, the key concern is not only the prevalence of CH, but also that CH-derived variants may occur in genes that would otherwise be interpreted as clinically meaningful if plasma were analyzed in isolation. The practical value of such classifiers therefore lies in reducing incorrect biological attribution at the point of reporting. In other words, the model is useful because it changes what a “positive” plasma result actually means[36].
This logic is already evident in perioperative and longitudinal ctDNA workflows. In the LUCID study, Gale et al. used a tumor-informed design based on exome sequencing of the primary tumor to build patient-specific assays, with matched normal blood used to remove leukocyte-derived variants before plasma tracking[9]. This should be viewed as more than a technical refinement. It is an acknowledgment that MRD interpretation becomes much less trustworthy if hematopoietic background is left unresolved from the start. Algorithmic CHIP removal combined with PBMC-based correction can likewise change which plasma calls are read as positive[35], suggesting that CH cannot be adequately addressed by a few fixed exclusion rules alone. The more likely direction is probabilistic filtering that uses broader variant-level context and matched-normal information when available. For perioperative NSCLC, that matters because ctDNA positivity is clinically useful only if it really reflects residual tumor burden, not background hematopoietic clonality.
Translating serial ctDNA into dynamic postoperative risk estimates
Before asking where AI changes this picture, it is worth fixing the clinical baseline against which any model must be judged. Gale et al. used highly sensitive, patient-specific RaDaR assays (up to 48 tumor-informed amplicons) to analyze 363 serial plasma samples from 88 patients with predominantly early-stage NSCLC[9]. ctDNA detection at the prespecified landmark (2 weeks to 4 months after treatment) occurred in 17% of patients and was strongly associated with shorter recurrence-free survival (RFS; HR 14.8) and overall survival (OS; HR 5.48), whereas ctDNA detected 1-3 days after surgery was not associated with recurrence. That contrast matters in itself: both the timing and the persistence of detectable ctDNA, not simply its presence, carry the prognostic weight. Serial sampling showed that ctDNA-positive samples preceded radiologic recurrence by a median of ~213 days, with most detectable tumor fractions below 0.1% and over one-third of samples below 0.01%, so even a well-designed tumor-informed landmark test still operates close to its analytical floor and leaves part of the biologically relevant disease unresolved at any single time point. This is the ceiling that any downstream model has to improve on, and it frames the problem as a longitudinal one rather than a single-threshold one.
Turning these serial trajectories into an explicit, decision-grade risk score is the step where AI is genuinely relevant. Wang et al. developed PRIME, an interpretable machine-learning model trained across six curative-intent NSCLC cohorts (493 patients with stage I-III disease treated with surgery or definitive chemoradiotherapy), with MRD assessed at a postoperative landmark 2 weeks to 4 months after treatment[52]. Rather than read ctDNA as a binary call, it combined landmark MRD with blood-based mutations in kelch-like ECH-associated protein 1 (KEAP1), serine/threonine kinase 11 (STK11) and cyclin-dependent kinase inhibitor 2A (CDKN2A) and with clinical features, after removal of germline and clonal-hematopoiesis variants. Of several algorithms, a neural network performed best, reaching an AUC of 0.85 (95%CI: 0.81-0.89) in training and 0.82 (0.74-0.89) in independent validation, with landmark MRD the dominant contributor in the SHAP analysis; model-defined high-risk patients derived a significant benefit from adjuvant therapy after surgery (P < 0.001), whereas low-risk patients did not (P = 0.928). For a perioperative reader, this is close to the intended task - a curative-intent cohort, a postoperative landmark, and an output that maps onto an adjuvant-therapy decision - but it is not a finished tool. The training population deliberately pooled resectable with unresectable and surgical with chemoradiotherapy patients, a heterogeneity the authors list first among their limitations, so its calibration in a strictly resectable, surgery-only setting remains to be shown, and the supporting evidence is still retrospective.
Zhang et al. approached the same problem from the side of the negative result[53]. Single-time-point landmark MRD is insensitive - its false-negative rate approaches 70%, and its sensitivity is only about 30% - so LAMPAD, an XGBoost-Cox model, pairs the postoperative landmark with a preoperative baseline draw to recover prognostic information from landmark-negative patients. Trained on 163 resected stage I-III patients (no neoadjuvant therapy) with undetectable landmark MRD and validated across cohorts using both fixed-panel and personalized assays, it separated low- from high-risk patients with 2-year disease-free survival of 97.8% vs. 71.6% (HR 0.11, 95%CI: 0.06-0.21), a separation that held in pooled validation (94.3% vs. 72.4%; HR 0.18, 95%CI: 0.13-0.25), with baseline ctDNA level as the leading feature; methylation deconvolution traced the high-risk signal to immune- and multi-organ-derived cfDNA rather than tumor shedding alone. As direct perioperative evidence, this is valuable because it targets the weak negative result that most limits clinical actionability and does so in precisely the resected population of interest. The same caveats apply: the report is in press, the dual-time-point design depends on a preoperative sample that is not always banked, the validation is retrospective, and the authors note that compatibility with fragmentomic and methylation-based platforms is unproven.
The clearest demonstrations of how longitudinal ctDNA trajectories can be translated into risk scores currently come from metastatic NSCLC, where dense serial sampling is available, and AI/ML methods have been applied at scale. Assaf et al. analyzed 466 patients from the randomized phase III IMpower150 trial, sampling ctDNA at baseline and through early on-treatment timepoints, and - after algorithmic and PBMC-based removal of germline and clonal-hematopoiesis variants - trained a machine-learning model that integrated longitudinal features such as changes in VAF and the number of detectable alterations over time to predict OS[35]. The resulting score separated patients with radiographic stable disease or partial response into high- versus low-intermediate-risk groups (hazard ratios ~3.2-3.3), with wide OS separations (for example, 7.1 months vs. 22.3 months in stable disease), and remained prognostic in an independent OAK validation cohort; in simulation, early ctDNA dynamics could even outperform early imaging for predicting trial-level OS differences. Yuan et al. reworked the same data into a treatment-agnostic, nonlinear mixed-effects joint model that links ctDNA kinetics to OS without using the treatment arm as a covariate, producing predicted survival rates generally within 20% - often within 10% - of the final trial readouts[54]. Both are methodologically instructive, and the joint-modeling logic in particular transfers naturally to a postoperative setting. The domain does not transfer with it, however: these models were built where tumor fractions are high and serial sampling is dense, whereas perioperative MRD operates near the limit of detection and on a different endpoint, so their figures are better treated as hypothesis-generating for the resectable setting[54].
Meta-analytic data reinforce the need to move beyond binary ctDNA endpoints when constructing such models. Lu et al. show that study-level performance improves when postoperative ctDNA is analyzed longitudinally, with AUCs approaching ~0.9 in several early-stage NSCLC subgroups[55]. Costa et al. further argue that ctDNA VAF dynamics capture subtle changes in tumor burden that may precede both radiologic progression and clinical deterioration, making them particularly amenable to AI/ML techniques that can compress high-frequency, low-amplitude signals into continuous risk scores[30]. Encouragingly, this is no longer a borrowed argument - the modeling is now being done in the resectable population it is meant to serve. What remains is the harder part, and it is not unique to AI: a risk score earns its place in perioperative care only once it has been tested prospectively, shown to hold across centers and assays, and tied to a decision a clinician would actually make differently. To make the evidentiary basis of this section explicit, Table 2 maps the AI/ML studies by their proximity to the target use case, separating direct perioperative NSCLC MRD evidence from findings extrapolated from screening, diagnostic, metastatic, or active-disease monitoring settings. A structured comparison of these AI/ML methods and validation features is provided in Supplementary Table 5.
Evidence gap map for AI/ML studies informing perioperative ctDNA-MRD in resectable NSCLC
| Evidence category | Study | Original context and domain gap | Transfer implication for perioperative NSCLC |
| Direct | Wang et al., 2023[49] | Resected NSCLC cohort with no neoadjuvant therapy; 87 patients, 163 plasma samples at 7 days and 6 months after curative surgery. Penalized Cox fragmentomic models were evaluated by leave-one-out cross-validation | Meets the Direct category because the evaluated population is resected NSCLC with postoperative MRD/recurrence endpoints. Evidence maturity remains limited by single-center leave-one-out validation, so thresholds and transportability need independent external validation |
| Zhang et al., 2026[53] | Stage I-III NSCLC patients undergoing surgical resection without neoadjuvant therapy; preoperative baseline and postoperative landmark blood were integrated with clinicopathologic variables for DFS/cure-risk stratification | Meets the Direct category because the development/evaluation setting is postoperative resected NSCLC. It provides multi-cohort AI/ML evidence, but prospective treatment-guidance testing is still needed | |
| Direct-adjacent | Widman et al., 2024[47] | Early-stage NSCLC arm from a neoadjuvant immunotherapy protocol: 22 patients received durvalumab with or without SBRT before intended surgery; 16 had postoperative plasma and 14 underwent resection. Postoperative MRD was associated with recurrence, but samples were drawn in a neoadjuvant/adjuvant-treated context | Direct-adjacent because the NSCLC evidence is a component of a mixed-cancer/platform study and was neoadjuvant-treated. Best used as neoadjuvant-treated perioperative evidence for MRD-EDGE sensitivity; broader postoperative NSCLC deployment requires separate validation |
| Zhu et al., 2025[50] | Multi-cancer deep-learning model for ctDNA quantification from fragment-length distributions; applied to a resected early-stage lung cancer cohort at the landmark timepoint about 30 days after curative surgery | Direct-adjacent because the lung MRD application is clinically close, but the primary model task is ctDNA quantification rather than a postoperative recurrence or treatment-decision model | |
| Zviran et al., 2020[48] | Genome-wide cfDNA MRD method paper with a real postoperative early-stage LUAD arm: 22 postoperative samples collected at a median of 2.5 weeks after surgery, within a mixed-cancer development study | Direct-adjacent feasibility evidence for ultra-sensitive genome-wide MRD detection, but limited by mixed cancer cohorts, small LUAD sample size, internal validation, and incomplete maturity for NSCLC deployment | |
| Wang et al., 2026[52] | Curative-intent stage I-III NSCLC model pooling radical surgery and definitive radiotherapy/chemoradiotherapy cohorts; treatment modality is one of the model predictors, with a resectable surgical subgroup reported | Direct-adjacent for localized NSCLC risk modeling, but not a pure postoperative resected-NSCLC MRD model. Surgical deployment requires subgroup-specific calibration and validation | |
| Metastatic/active-disease | Assaf et al., 2023[35] | Metastatic non-squamous NSCLC from IMpower150; longitudinal on-treatment ctDNA dynamics were modeled across five timepoints to predict overall survival | Useful as a mature longitudinal modeling template, but metastatic tumor burden, sampling density, active systemic therapy, and OS endpoints do not transfer directly to low-burden postoperative MRD |
| Yuan et al., 2025[54] | Treatment-agnostic joint model of longitudinal ctDNA dynamics in treatment-naive metastatic non-squamous NSCLC from IMpower150, predicting overall survival across first-line regimens | The joint-modeling structure is informative, but any perioperative use would require refitting and validation in true post-resection cohorts with recurrence/DFS endpoints | |
| van ’t Erve et al., 2024[73] | Fragmentome-based tumor-fraction estimator developed mainly in metastatic CRC and validated for longitudinal treatment monitoring, with an independent stage III/IV lung cancer validation cohort | Supports mutation-independent fragmentomic tumor-fraction estimation as a feature class, but active-disease monitoring does not establish postoperative NSCLC MRD performance | |
| Screening/diagnosis | Bahado-Singh et al., 2022[45] | Small lung-cancer-versus-control methylation study using supervised and deep-learning classifiers; n = 30 with near-perfect reported discrimination | Mechanistically relevant for methylation feature integration, but the task is prevalent cancer detection. It should be framed as proof-of-concept and an overfitting caution, not MRD evidence |
| Kim et al., 2024[46] | NSCLC diagnosis versus healthy controls using a CNN over cfDNA methylation and fragment-size profiles; includes dilution experiments but not postoperative recurrence prediction | Supports joint methylation-fragment modeling for weak signal recovery, but postoperative operating points, specificity, and false-positive tolerance must be re-established | |
| Mathios et al., 2021[71] | Prospective lung cancer detection and characterization from cfDNA fragmentomes in high-risk/symptomatic cohorts, validated against non-cancer individuals and lung cancer cases | Supports genome-wide fragmentomic features and external validation practice; detection of prevalent cancer is not equivalent to ultra-low postoperative residual disease | |
| Mazzone et al., 2024[72] | Prospective case-control validation of a locked low-coverage WGS fragmentome classifier for blood-based lung cancer screening among screening-eligible individuals | Shows that fragmentome classifiers can be clinically validated, but screening sensitivity/specificity trade-offs differ from perioperative MRD decision thresholds | |
| CH filtering | Fairchild et al., 2023[36] | Pan-cancer plasma-only classifier distinguishing blood-derived clonal hematopoiesis variants from tumor-derived variants in cfDNA sequencing | Relevant for improving MRD specificity and reducing false positives, but it is a variant-origin task rather than a postoperative recurrence-risk model |
| Arango-Argoty et al., 2025[62] | Open-source ML framework that classifies cfDNA variants as CH or tumor origin from plasma-only samples in the absence of matched WBC sequencing | Useful when matched-WBC sequencing is unavailable, but perioperative NSCLC deployment should be benchmarked against matched-WBC labels and outcome-aware ablation tests | |
| Palizban et al., 2024[63] | Semi-supervised GAN model for classifying somatic mutation origins in cfDNA as CH-related or tumor-derived using curated variant-origin labels | Mainly informs specificity control and transfer-risk discussion; high internal AI performance in variant attribution does not establish MRD recurrence prediction |
CHALLENGES FOR AI-ENABLED ctDNA-MRD IN PERIOPERATIVE NSCLC
Window and threshold drift
In perioperative NSCLC, the clinical meaning of a post-treatment MRD result depends strongly on when the blood is drawn[9], and any model built on these data will also be constrained by the sensitivity of the underlying assay. An early postoperative landmark sample and a later surveillance sample should not be assumed to represent the same clinical context[32,56]. These differences suggest that indiscriminate pooling of postoperative and surveillance windows may complicate model development and interpretation. A mixed-window model may appear acceptable overall while still being less informative within specific clinical windows. A more conservative and better-supported conclusion is that predictive performance differs across postoperative windows and monitoring strategies. In localized NSCLC, landmark MRD detection yielded an NPV/PPV of 86.6%/81.0%, which increased to 96.8%/89.1% when longitudinal time points were incorporated[32]. Existing perioperative NSCLC data support this interpretation. In resected NSCLC, postoperative sampling windows vary substantially across studies, ranging from a few days to several months, and landmark and surveillance strategies show different performance profiles, with surveillance generally improving sensitivity while changing the underlying prevalence structure of the task[10,24]. These findings argue against treating sampling time as a purely incidental feature in perioperative MRD modeling. A more defensible approach may be to prespecify the intended clinical windows and evaluate model behavior separately within them, rather than assuming that one post-treatment setting is interchangeable with another. At a minimum, separating early postoperative assessment from later surveillance may be more clinically interpretable than applying a single cutoff across the entire perioperative course.
CH confounding
At the ultra-low variant allele fractions encountered in MRD analysis, the interpretation of plasma variants can be substantially confounded by CH[51,57,58], especially in older patients, smokers, or previously treated patients[34,59]. If plasma variants are treated indiscriminately as tumor-derived, model outputs may partly reflect CH-associated background rather than residual NSCLC alone[60]. This distinction matters because, when CH is left unresolved, the model may no longer be answering a clean perioperative MRD question. Instead, the task may shift from “is residual NSCLC present?” to “does this patient have residual NSCLC, or a plasma background enriched for CH?” In a plasma-only pipeline, those are not the same problem. When feasible, matched white-blood-cell or PBMC sequencing remains the most robust design choice[34,51,58,61]. When it cannot, CH handling should be described explicitly rather than noted only in general terms. In such cases, an explicitly described and independently evaluated in silico CH-filtering strategy is preferable to an unspecified adjustment[62,63]. Just as importantly, perioperative AI studies would also benefit from showing how model performance changes when CH handling is omitted. Ablation analyses with and without CH filtering may be informative, because they make it possible to see whether apparent model performance is being driven by residual disease biology or by hematopoietic background. Specificity and PPV should therefore be reported transparently, especially when background non-tumor signal may influence classification. Otherwise, a model may appear accurate while assigning molecular risk on an incorrect biological basis.
Assay and site effects
Fragmentomic MRD features are influenced by both biological and technical context. Their observed values can be shaped by both biology and the analytical pipeline, although the size of this effect is feature-specific rather than universal[64-66]. In cfDNA fragmentomics, preanalytical and analytical factors such as collection and processing conditions, library preparation, and bioinformatic processing can alter some readouts, particularly genome-wide fragmentation patterns and fragment-end features[66]. By contrast, not every variable perturbs every feature to the same degree; for example, collection tube and processing time did not materially change overall fragment-size distributions in one study, whereas genome-wide fragmentation patterns and fragment-end sequences were affected, and one- versus two-step centrifugation had little effect on nuclear cfDNA size or motif diversity but did affect mitochondrial cfDNA[65]. Wang et al. made this problem especially clear by comparing donor-derived plasma cfDNA processed with nine library kits and ten data-processing routes: kit- and route-dependent variation was evident, particularly for motif profiles and some regional length signals, and batch effects remained visible across published datasets even after standardized reprocessing[66]. In perioperative NSCLC, where post-treatment ctDNA can be present at extremely low fractions, such technical shifts cannot be dismissed, because assay-dependent variation may be mistaken for weak biological signal[33]. This does not imply that every strong single-center model is non-portable, but it does indicate that portability should not be inferred from internal performance alone[67,68]. Cross-study fragmentomics work has specifically highlighted the need for external validation, and claims of generalizability are better supported when models are evaluated in independent site-held-out data. For reporting, the most defensible standard is to describe the assay and preprocessing pipeline in enough detail for reproduction, identify the development and validation settings, and report discrimination and calibration transparently, including any heterogeneity across clinical windows or centers[69,70]. By contrast, pipeline locking, version control, and longitudinal drift monitoring are best framed as strong translational recommendations rather than as conclusions already established by perioperative NSCLC MRD studies.
Domain shift
Many published fragmentomic AI models for lung cancer have been trained in screening, diagnostic, or active-disease treatment-monitoring cohorts rather than in postoperative MRD cohorts[71-73]. Models developed directly within resected, postoperative NSCLC populations remain few, and the existing examples are typically limited by small cohort size and an absence of independent validation[49]. Perioperative MRD in resectable NSCLC belongs to a different domain, with post-treatment ctDNA often approaching the analytical limits of detection and the task shifting from current cancer detection to future recurrence prediction. For this reason, perioperative AI-MRD models should ideally be developed in genuine perioperative cohorts, or at minimum externally validated and, when necessary, recalibrated in such cohorts, rather than transferred unchanged from screening or active-disease settings[74]. The target population, outcome definition, and signal-to-noise regime are different, and postoperative samples generally show a much weaker tumor signal than cancer-versus-control or active-disease settings. A model that behaves sensibly in a “cancer versus control” setting may therefore become poorly calibrated once it is moved into a postoperative relapse-prediction setting[67,72]. In practice, this means that recalibration and threshold re-estimation may be needed after external validation, especially when models are moved across clinical windows or domains.
Risk-action gap
In perioperative NSCLC, an MRD score becomes clinically useful only when it is linked to a prespecified clinical action. A probability estimate alone is insufficient. This is one reason why strong retrospective performance does not automatically translate into practice[31,75-77]. The postoperative MRD literature in NSCLC remains heterogeneous, there is still no single standard-of-care framework for how liquid-biopsy MRD should be deployed, and recent meta-analytic work continues to show that performance depends on assay type, monitoring strategy, and the clinical context in which the test is applied[78-80]. Thus, the central task is not only to build a model that ranks risk, but also to define what action should follow each risk level. One practical route is to align the score with the target perioperative population and map the output into a small number of prespecified action categories: closer surveillance, continuation of standard therapy, consideration of escalation, or in selected contexts de-escalation[81]. The goal is not to force every score into an intervention. It is to prevent the opposite problem, where the same model output may be interpreted differently across centers because no action framework was set in advance. Without that calibration-plus-action structure, even a statistically strong MRD model is likely to generate variable reporting, variable management, and very little workflow benefit[31]. In other words, the model may predict risk, but it will not support reproducible care unless its outputs are linked to predefined clinical actions[75].
The major sources of uncertainty and potential AI/ML failure modes across the perioperative ctDNA-MRD workflow are summarized in Figure 2.
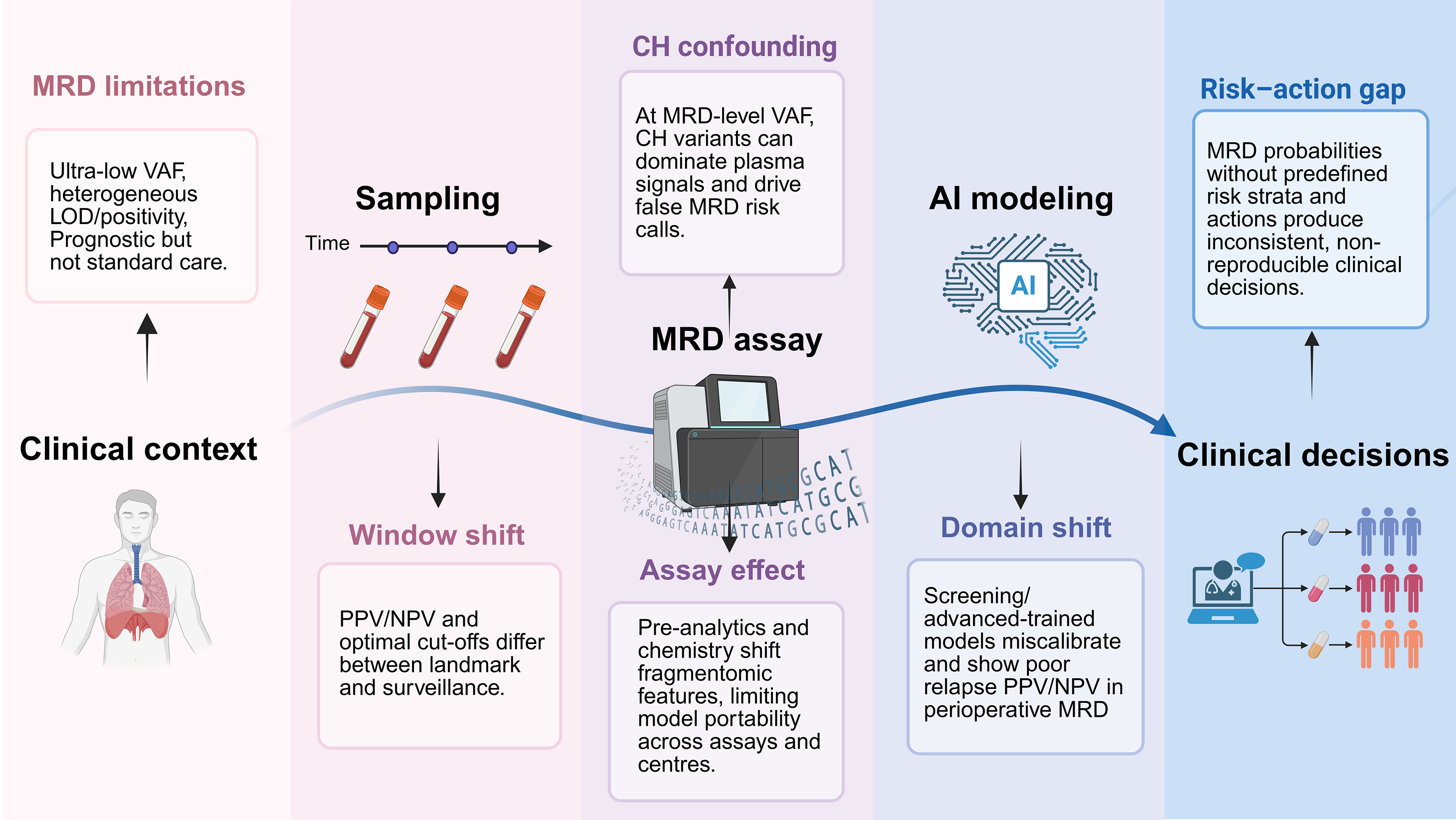
Figure 2. Perioperative ctDNA-MRD workflow in resectable NSCLC and AI/ML failure modes across the pipeline. The central curve depicts the perioperative ctDNA-MRD workflow, from clinical context and sampling to MRD assay, AI modeling, and clinical decision-making. The color-coded panels map MRD-intrinsic constraints (MRD limitations, CH confounding) and AI/ML-specific failure modes (window shift, assay effect, domain shift, risk-action gap) to each pipeline layer. Created in BioRender. Chen, Y. (2026) https://BioRender.com/tvny9l2. MRD: Minimal residual disease; ctDNA: circulating tumor DNA; VAF: variant allele fraction; LOD: limit of detection; CH: clonal hematopoiesis; AI: artificial intelligence; ML: machine learning; PPV: positive predictive value; NPV: negative predictive value.
FUTURE DIRECTIONS
The next phase of perioperative ctDNA-MRD research in resectable NSCLC should move beyond retrospective demonstrations that postoperative ctDNA positivity is associated with recurrence. This association has been consistently demonstrated. The more important question is now practical: which patients should actually enter prospective treatment-guiding trials, and which MRD patterns are strong enough to justify changing management. Future studies need to test this directly. Persistent MRD positivity at a clinically meaningful postoperative landmark, failure of ctDNA clearance, or conversion from negative to positive during surveillance are all plausible candidates for treatment intensification. The converse question is just as important and probably harder: whether sustained MRD negativity across predefined time windows can support shorter adjuvant therapy, de-escalation, or even treatment omission in carefully selected patients. A clinically meaningful advance will require linking MRD-defined risk groups to prespecified interventions and evaluating them using clinically relevant outcomes, rather than relying only on separation of Kaplan-Meier curves. Relevant endpoints should include recurrence-free survival, reduction of overtreatment, and the safety of withholding or shortening therapy, rather than relying on indirect inference.
AI is most relevant when it is applied to interpretive tasks that conventional approaches do not handle well. In perioperative NSCLC, three applications appear particularly relevant. The first is longitudinal risk modeling from serial ctDNA measurements, so that recurrence risk can be updated over time rather than reduced to a single positive-or-negative result. Although direct evidence remains limited, future AI-based models could explore whether integrating ctDNA clearance dynamics with postoperative ctDNA status improves recurrence-risk estimation after neoadjuvant therapy. Such models could provide a more comprehensive assessment of treatment response and residual disease burden, potentially improving recurrence-risk prediction following neoadjuvant therapy. The second is calibration across clinically different sampling windows, because a ctDNA result does not mean the same thing immediately after surgery, at a postoperative landmark draw, or months later during surveillance. The third is variant interpretation in the presence of CH and assay-specific background noise, which becomes more problematic as tumor fraction approaches the lower limit of detection. These applications should therefore be framed not as generic opportunities to “apply AI,” but as defined perioperative tasks. They should be evaluated against management-relevant endpoints and decisions, not mainly against discrimination statistics in development datasets.
For ctDNA-MRD tools to be used credibly in practice, improved model performance alone will not be sufficient. Algorithms should be prespecified before prospective testing, calibrated in the perioperative population in which it is meant to be used, and then shown to hold up across centers, assays, and sampling workflows. Just as importantly, the output has to connect to a limited set of real management options: escalate treatment, continue standard adjuvant therapy, step treatment down, or intensify surveillance. Otherwise the model is only producing a risk score in the abstract. Reporting standards should also be rigorous. Sampling window, assay context, CH handling, calibration, site-held-out validation, and the thresholds used to support treatment decisions all need to be explicit. Without that level of structure, AI-enabled ctDNA-MRD is likely to remain a sophisticated prognostic layer rather than a tool that can consistently shape perioperative care in NSCLC.
CONCLUSION
In resectable NSCLC, the central challenge is not simply detecting recurrence, but identifying residual disease early and reliably enough to inform perioperative and postoperative management. ctDNA-based MRD has therefore emerged as a clinically relevant adjunct for risk stratification, relapse surveillance, and assessment of perioperative treatment response. Its current role, however, remains constrained by limited sensitivity in low-shedding disease, imperfect specificity related to biological background signals such as CH, interpretive uncertainty across clinical contexts, and the lack of standardized analytical and reporting frameworks.
In this setting, AI is relevant because it may strengthen weak-signal detection, improve variant-origin assignment, and convert serial ctDNA measurements into more clinically interpretable risk estimates. These advances will become clinically meaningful only if they are supported by prospective validation, robust standardization, and clear integration into treatment and surveillance pathways. The continued refinement of ctDNA-based MRD technologies, together with more standardized workflows and AI-enabled interpretation, may help move perioperative NSCLC management toward more reliable and clinically actionable precision care.
DECLARATIONS
Acknowledgments
The Graphical Abstract was created in BioRender. Chen, Y. (2026) https://BioRender.com/p5kiylk.
Authors’ contributions
Conceptualization:
Literature search and data collection: Chen Y, Li W, Qiu
Drafting of the manuscript: Chen Y, Li
Preparation of tables and supplementary materials: He M, Chen
Figure design and visualization: Chen Y, Li
Critical revision for important intellectual content: Zhong
Supervision: Liang
All authors read and approved the final manuscript.
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
During the preparation of this manuscript, the AI tool ChatGPT (version GPT-5.4, released 2026-03-05) was used solely for language editing. The tool did not influence the study design, data collection, analysis, interpretation, or the scientific content of the work. All authors take full responsibility for the accuracy, integrity, and final content of the manuscript.
Financial support and sponsorship
None.
Conflicts of interest
He
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
Supplementary Materials
REFERENCES
1. Bray F, Laversanne M, Sung H, et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2024;74:229-63.
2. Rajaram R, Huang Q, Li RZ, et al. Recurrence-free survival in patients with surgically resected non-small cell lung cancer. Chest. 2024;165:1260-70.
3. Uramoto H, Tanaka F. Recurrence after surgery in patients with NSCLC. Transl Lung Cancer Res. 2014;3:242-9.
4. Fonseca NM, Maurice-Dror C, Herberts C, et al. Prediction of plasma ctDNA fraction and prognostic implications of liquid biopsy in advanced prostate cancer. Nat Commun. 2024;15:1828.
5. Garcia-Murillas I, Chopra N, Comino-Méndez I, et al. Assessment of molecular relapse detection in early-stage breast cancer. JAMA Oncol. 2019;5:1473-8.
6. Parikh AR, Van Seventer EE, Siravegna G, et al. Minimal Residual disease detection using a plasma-only circulating tumor DNA assay in patients with colorectal cancer. Clin Cancer Res. 2021;27:5586-94.
7. Sfakianos JP, Basu A, Laliotis G, et al. Association of tumor-informed circulating tumor DNA detectability before and after radical cystectomy with disease-free survival in patients with bladder cancer. Eur Urol Oncol. 2025;8:306-14.
8. Kurma K, Eslami-S Z, Alix-Panabières C, Cayrefourcq L. Liquid biopsy: paving a new avenue for cancer research. Cell Adh Migr. 2024;18:1-26.
9. Gale D, Heider K, Ruiz-Valdepenas A, et al. Residual ctDNA after treatment predicts early relapse in patients with early-stage non-small cell lung cancer. Ann Oncol. 2022;33:500-10.
10. Qiu B, Guo W, Zhang F, et al. Dynamic recurrence risk and adjuvant chemotherapy benefit prediction by ctDNA in resected NSCLC. Nat Commun. 2021;12:6770.
11. Abbosh C, Frankell AM, Harrison T, et al. ; TRACERx Consortium. Tracking early lung cancer metastatic dissemination in TRACERx using ctDNA. Nature. 2023;616:553-62.
12. Herbst RS, John T, Grohé C, et al. Molecular residual disease analysis of adjuvant osimertinib in resected EGFR-mutated stage IB-IIIA non-small-cell lung cancer. Nat Med. 2025;31:1958-68.
13. Abbosh C, Birkbak NJ, Wilson GA, et al. ; PEACE consortium. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature. 2017;545:446-51.
14. Hong TH, Hwang S, Dasgupta A, et al. Clinical utility of tumor-naïve presurgical circulating tumor DNA detection in early-stage NSCLC. J Thorac Oncol. 2024;19:1512-24.
15. Gassa A, Fassunke J, Schueten S, et al. Detection of circulating tumor DNA by digital droplet PCR in resectable lung cancer as a predictive tool for recurrence. Lung Cancer. 2021;151:91-6.
16. Chaudhuri AA, Chabon JJ, Lovejoy AF, et al. Early detection of molecular residual disease in localized lung cancer by circulating tumor DNA profiling. Cancer Discov. 2017;7:1394-403.
17. Newman AM, Lovejoy AF, Klass DM, et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat Biotechnol. 2016;34:547-55.
18. Kurtz DM, Soo J, Co Ting Keh L, et al. Enhanced detection of minimal residual disease by targeted sequencing of phased variants in circulating tumor DNA. Nat Biotechnol. 2021;39:1537-47.
19. Normanno N, Morabito A, Rachiglio AM, et al. Circulating tumour DNA in early stage and locally advanced NSCLC: ready for clinical implementation? Nat Rev Clin Oncol. 2025;22:215-31.
20. Peng M, Huang Q, Yin W, et al. Circulating Tumor DNA as a prognostic biomarker in localized non-small cell lung cancer. Front Oncol. 2020;10:561598.
21. Waldeck S, Mitschke J, Wiesemann S, et al. Early assessment of circulating tumor DNA after curative‐intent resection predicts tumor recurrence in early-stage and locally advanced non‐small‐cell lung cancer. Mol Oncol. 2021;16:527-37.
22. Zhang JT, Liu SY, Gao W, et al. Longitudinal undetectable molecular residual disease defines potentially cured population in localized non-small cell lung cancer. Cancer Discov. 2022;12:1690-701.
23. Xia L, Mei J, Kang R, et al. Perioperative ctDNA-based molecular residual disease detection for non-small cell lung cancer: a prospective multicenter cohort study (LUNGCA-1). Clin Cancer Res. 2022;28:3308-17.
24. Chen K, Zhao H, Shi Y, et al. Perioperative dynamic changes in circulating tumor DNA in patients with lung cancer (DYNAMIC). Clin Cancer Res. 2019;25:7058-67.
25. Felip E, Srivastava M, Reck M, et al. 1O IMpower010: ctDNA status in patients (pts) with resected NSCLC who received adjuvant chemotherapy (chemo) followed by atezolizumab (atezo) or best supportive care (BSC). Immuno-Oncol. Technol. 2022;16:100106.
26. Provencio M, Serna-Blasco R, Nadal E, et al. Overall survival and biomarker analysis of neoadjuvant nivolumab plus chemotherapy in operable stage IIIA non-small-cell lung cancer (NADIM phase II trial). J Clin Oncol. 2022;40:2924-33.
27. Reck M, Gale D, Harpole D, et al. LBA59 Associations of ctDNA clearance and pathological response with neoadjuvant treatment in patients with resectable NSCLC from the phase III AEGEAN trial. Ann Oncol. 2023;34:S1300.
28. Reck M, Gale D, Zhu Z, et al. LBA49 Associations of ctDNA clearance (CL) during neoadjuvant Tx with pathological response and event-free survival (EFS) in pts with resectable NSCLC (R-NSCLC): expanded analyses from AEGEAN. Ann Oncol. 2024;35:S1239.
29. Yue D, Liu W, Chen C, et al. Circulating tumor DNA predicts neoadjuvant immunotherapy efficacy and recurrence-free survival in surgical non-small cell lung cancer patients. Transl Lung Cancer Res. 2022;11:263-76.
30. Costa J, Membrino A, Zanchetta C, et al. The role of ctDNA in the management of non-small-cell lung cancer in the AI and NGS era. Int J Mol Sci. 2024;25:13669.
31. Boukouris AE, Michaelidou K, Joosse SA, et al. A comprehensive overview of minimal residual disease in the management of early-stage and locally advanced non-small cell lung cancer. NPJ Precis Oncol. 2025;9:178.
32. Zhong R, Gao R, Fu W, et al. Accuracy of minimal residual disease detection by circulating tumor DNA profiling in lung cancer: a meta-analysis. BMC Med. 2023;21:180.
33. Pellini B, Chaudhuri AA. Circulating tumor DNA minimal residual disease detection of non-small-cell lung cancer treated with curative intent. J Clin Oncol. 2022;40:567-75.
34. Yaung SJ, Fuhlbrück F, Peterson M, et al. Clonal hematopoiesis in late-stage non-small-cell lung cancer and its impact on targeted panel next-generation sequencing. JCO Precis Oncol. 2020:1271-9.
35. Assaf ZJF, Zou W, Fine AD, et al. A longitudinal circulating tumor DNA-based model associated with survival in metastatic non-small-cell lung cancer. Nat Med. 2023;29:859-68.
36. Fairchild L, Whalen J, D’Aco K, et al. Clonal hematopoiesis detection in patients with cancer using cell-free DNA sequencing. Sci Transl Med. 2023;15:eabm8729.
37. Bestvina CM, Garassino MC, Neal JW, Wakelee HA, Diehn M, Vokes EE. Early-stage lung cancer: using circulating tumor DNA to get personal. J Clin Oncol. 2023;41:4093-6.
38. Olmedillas-López S, García-Arranz M, García-Olmo D. Current and emerging applications of droplet digital PCR in oncology. Mol Diagn Ther. 2017;21:493-510.
39. Wu LR, Chen SX, Wu Y, Patel AA, Zhang DY. Multiplexed enrichment of rare DNA variants via sequence-selective and temperature-robust amplification. Nat Biomed Eng. 2017;1:714-23.
40. Cascone T, Awad MM, Spicer JD, et al. Perioperative nivolumab in resectable lung cancer. N Engl J Med. 2024:390:1756-69.
41. Forde PM, Spicer J, Lu S, et al. Neoadjuvant nivolumab plus chemotherapy in resectable lung cancer. N Engl J Med. 2022;386:1973-85.
42. Forde PM, Spicer JD, Provencio M, et al. Overall survival with neoadjuvant nivolumab plus chemotherapy in lung cancer. N Engl J Med. 2025;393:741-52.
43. Deveson IW, Gong B, Lai K, et al. ; SEQC2 Oncopanel Sequencing Working Group. Evaluating the analytical validity of circulating tumor DNA sequencing assays for precision oncology. Nat Biotechnol. 2021;39:1115-28.
44. Thalambedu N, Balla M, Sivasubramanian BP, et al. Integrating artificial intelligence with circulating tumor DNA for non-small cell lung cancer: opportunities, challenges, and future directions. Front Med. 2025;12:1612376.
45. Bahado-Singh R, Vlachos KT, Aydas B, Gordevicius J, Radhakrishna U, Vishweswaraiah S. Precision oncology: artificial intelligence and DNA methylation analysis of circulating cell-free DNA for lung cancer detection. Front Oncol. 2022;12:790645.
46. Kim M, Park J, Seonghee Oh , et al. Deep learning model integrating cfDNA methylation and fragment size profiles for lung cancer diagnosis. Sci Rep. 2024;14:14797.
47. Widman AJ, Shah M, Frydendahl A, et al. Ultrasensitive plasma-based monitoring of tumor burden using machine-learning-guided signal enrichment. Nat Med. 2024;30:1655-66.
48. Zviran A, Schulman RC, Shah M, et al. Genome-wide cell-free DNA mutational integration enables ultra-sensitive cancer monitoring. Nat Med. 2020;26:1114-24.
49. Wang S, Xia Z, You J, et al. Enhanced detection of landmark minimal residual disease in lung cancer using cell-free DNA fragmentomics. Cancer Res Commun. 2023;3:933-42.
50. Zhu G, Rahman CR, Getty V, et al. A deep-learning model for quantifying circulating tumour DNA from the density distribution of DNA-fragment lengths. Nat Biomed Eng. 2025;9:307-19.
51. Razavi P, Li BT, Brown DN, et al. High-intensity sequencing reveals the sources of plasma circulating cell-free DNA variants. Nat Med. 2019;25:1928-37.
52. Wang Y, Xiang YB, Chen XW, et al. PRIME: an interpretable artificial intelligence model based on liquid biopsy improves prediction of progression risk in non-small cell lung cancer. Mil Med Res. 2026;12:94.
53. Zhang JT, Chen KZ, Gao X, et al. LAMPAD: an integrated circulating tumor DNA-based model for predicting potential cure in patients with resected NSCLC. J Thorac Oncol. 2026:103729.
54. Yuan M, Lin X, Ding H, Qu S, Yang Y, Steven Xu X. Treatment-agnostic joint modeling of longitudinal circulating tumor dna predicts survival across first-line regimens in metastatic non-squamous NSCLC. Eur J Pharm Sci. 2025;214:107275.
55. Lu D, Lin N, Li S, et al. Predictive effectiveness of circulating tumor DNA in recurrent early-stage non-small cell lung cancer: an updated meta-analysis. JCO Precis Oncol. 2025;9:e2500489.
56. Schuurbiers MMF, Smith CG, Hartemink KJ, et al. ; the LEMA Study Group and the LUCID Study Group. Recurrence prediction using circulating tumor DNA in patients with early-stage non-small cell lung cancer after treatment with curative intent: a retrospective validation study. PLoS Med. 2025;22:e1004574.
57. Abbosh C, Birkbak NJ, Swanton C. Early stage NSCLC - challenges to implementing ctDNA-based screening and MRD detection. Nat Rev Clin Oncol. 2018;15:577-86.
58. Hu Y, Ulrich BC, Supplee J, et al. False-positive plasma genotyping due to clonal hematopoiesis. Clin Cancer Res. 2018;24:4437-43.
59. Coombs CC, Zehir A, Devlin SM, et al. Therapy-related clonal hematopoiesis in patients with non-hematologic cancers is common and associated with adverse clinical outcomes. Cell Stem Cell. 2017;21:374-82.e4.
60. Yun JK, Kim S, An H, et al. Pre-operative clonal hematopoiesis is related to adverse outcome in lung cancer after adjuvant therapy. Genome Med. 2023;15:111.
61. Leal A, van Grieken NCT, Palsgrove DN, et al. White blood cell and cell-free DNA analyses for detection of residual disease in gastric cancer. Nat Commun. 2020;11:525.
62. Arango-Argoty G, Haghighi M, Sun GJ, et al. An artificial intelligence-based model for prediction of clonal hematopoiesis variants in cell-free DNA samples. NPJ Precis Oncol. 2025;9:147.
63. Palizban F, Sarbishegi M, Kavousi K, Mehrmohamadi M. Predicting somatic mutation origins in cell-free DNA by semi-supervised GAN models. Heliyon. 2024;10:e39379.
64. van der Pol Y, Moldovan N, Verkuijlen S, et al. The effect of preanalytical and physiological variables on cell-free DNA fragmentation. Clin Chem. 2022;68:803-13.
65. Hu X, Zhang H, Wang Y, et al. Effects of blood-processing protocols on cell-free DNA fragmentomics in plasma: comparisons of one- and two-step centrifugations. Clin Chim Acta. 2024;560:119729.
66. Wang H, Mennea PD, Chan YKE, et al. A standardized framework for robust fragmentomic feature extraction from cell-free DNA sequencing data. Genome Biol. 2025;26:141.
67. van Calster B, Steyerberg EW, Wynants L, van Smeden M. There is no such thing as a validated prediction model. BMC Med. 2023;21:70.
68. Su S, Xuan Y, Fan X, et al. Testing the generalizability of cfDNA fragmentomic features across different studies for cancer early detection. Genomics. 2023;115:110662.
69. Debray TPA, Collins GS, Riley RD, et al. Transparent reporting of multivariable prediction models developed or validated using clustered data: TRIPOD-cluster checklist. BMJ. 2023;380:e071018.
70. Collins GS, Moons KGM, Dhiman P, et al. TRIPOD+AI statement: updated guidance for reporting clinical prediction models that use regression or machine learning methods. BMJ. 2024;385:e078378.
71. Mathios D, Johansen JS, Cristiano S, et al. Detection and characterization of lung cancer using cell-free DNA fragmentomes. Nat Commun. 2021;12:5060.
72. Mazzone PJ, Bach PB, Carey J, et al. Clinical validation of a cell-free DNA fragmentome assay for augmentation of lung cancer early detection. Cancer Discov. 2024;14:2224-42.
73. van ’t Erve I, Alipanahi B, Lumbard K, et al. Cancer treatment monitoring using cell-free DNA fragmentomes. Nat Commun. 2024;15:8801.
74. Riley RD, Archer L, Snell KIE, et al. Evaluation of clinical prediction models (part 2): how to undertake an external validation study. BMJ. 2024;384:e074820.
75. Kobayashi S, Nakamura Y, Hashimoto T, et al. Japan society of clinical oncology position paper on appropriate clinical use of molecular residual disease (MRD) testing. Int J Clin Oncol. 2025;30:605-54.
76. Sato Y. Liquid biopsy for minimal residual disease and monitoring in early-stage non-small cell lung cancer: current clinical utility and implementation challenges. Explor Med. 2025;6:1001349.
77. Denis MG. Molecular minimal residual disease in resected non-small cell lung cancer: “flattening the curve”. Transl Lung Cancer Res. 2025;14:4180-3.
78. Verzè M, Pluchino M, Leonetti A, et al. Role of ctDNA for the detection of minimal residual disease in resected non-small cell lung cancer: a systematic review. Transl Lung Cancer Res. 2022;11:2588-600.
79. Shi Y, Chen R, Xiong Y, et al. MRD status combined with TNM staging optimizes postoperative prognostic stratification in non-small cell lung cancer: a meta-analysis and systematic review. Lung Cancer. 2025;206:108634.
80. Ohara S, Suda K, Sudhaman S, et al. Clinical significance of perioperative minimal residual disease detected by circulating tumor DNA in patients with lung cancer with a long follow-up data: an exploratory study. JTO Clin Res Rep. 2025;6:100762.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].