


Histone lactylation in hepatocellular carcinoma: a key epigenetic hub linking metabolic reprogramming and the immune microenvironment
 0
0 
Abstract
Hepatocellular carcinoma (HCC) is a malignant tumor with extremely high global morbidity and mortality. Its initiation and progression are tightly associated with the complex tumor microenvironment (TME), which drives immune escape and treatment resistance in liver cancer. In recent years, epigenetic regulation, particularly histone modification mediated by cellular metabolism, has emerged as a critical advance in deciphering tumor malignant behavior and its interaction with the microenvironment. Histone lactylation, a newly identified post-translational modification directly mediated by lactate, offers a novel perspective for understanding how abnormal tumor metabolism (especially the Warburg effect) shapes the immune microenvironment via epigenetic reprogramming. This review elaborates on the role of histone lactylation in HCC progression, focusing on its mechanisms in regulating oncogenic signaling pathways, metabolic reprogramming and reshaping the HCC TME, thereby serving as a core node in metabolic-epigenetic-immune crosstalk. In addition, we summarize the intervention strategies and drug development targeting this modification, and discuss future directions, aiming to provide a basis for the development of novel HCC therapeutic strategies based on metabolic-epigenetic regulation.
Keywords
INTRODUCTION
Epigenetics refers to heritable changes that affect gene expression without modifying the DNA sequence itself, through DNA methylation, histone modification, chromatin remodeling, and non-coding RNA regulation[1-3]. These modifications are reversible and dynamic, enabling cells to respond to intracellular and extracellular environmental changes and thus participate in processes such as cell differentiation, development, aging, and disease pathogenesis[1-3]. Epigenetic dysregulation is a hallmark of tumor cells, characterized by hypermethylation-induced silencing of tumor suppressor genes, hypomethylation-driven activation of oncogenes, and aberrant histone modification patterns[2,4]. In eukaryotic chromatin, core histones undergo various post-translational modifications, forming a “histone code” that regulates chromatin accessibility by recruiting different effector protein complexes, thereby influencing the binding of transcription factors and gene expression[5,6]. Common histone modifications include acetylation, methylation, phosphorylation, and ubiquitination. For instance, histone acetylation, catalyzed by histone acetyltransferases (HATs), generally promotes gene transcription; in contrast, histone deacetylases (HDACs) remove acetyl groups, condensing chromatin and inhibiting gene expression[5,6]. While classic histone modifications (e.g., acetylation, methylation) have been extensively studied in hepatocellular carcinoma (HCC), how the unique metabolic state of tumor cells is directly converted into epigenetic instructions remains unclear.
Lactate was long considered a metabolic waste product of anaerobic glycolysis. However, the widespread “Warburg effect” in tumor cells, in which tumors preferentially produce large amounts of lactate via anaerobic glycolysis even in the presence of sufficient oxygen, creates an acidic tumor microenvironment (TME)[7,8]. Recently, studies have redefined lactate as a key signaling molecule and metabolic intermediate that regulates multiple cellular biological processes[9,10]. In 2019, Zhang et al. first discovered that lactate serves as a donor for histone modification, mediating lactylation modification on histone lysine residues (i.e., histone lactylation). This modification was shown to promote the expression of anti-inflammatory genes [e.g., interleukin (IL)-10], thereby regulating macrophage polarization[11]. This finding uncovered a new role of lactate in epigenetic regulation, established a direct link between cellular metabolism and gene expression, and provided a novel framework for understanding metabolic reprogramming in diseases[12,13]. Subsequently, non-histone protein lactylation has been identified and widely involved in a broad range of biological functions and disease development. With the rapid advances of mechanisms and functions of histone and non-histone lactylation, the understanding of this metabolic-epigenetic regulation has been greatly clarified (see review[12,14]). In brief, histone lactylation occurs on histones and regulates gene transcription by altering chromatin structure, representing a typical epigenetic regulatory mechanism. By contrast, non-histone lactylation targets non-histone proteins such as enzymes, signaling proteins, and transcription factors, and directly modulates protein stability, activity, subcellular localization, or molecular interactions, belonging to the functional regulation of proteins.
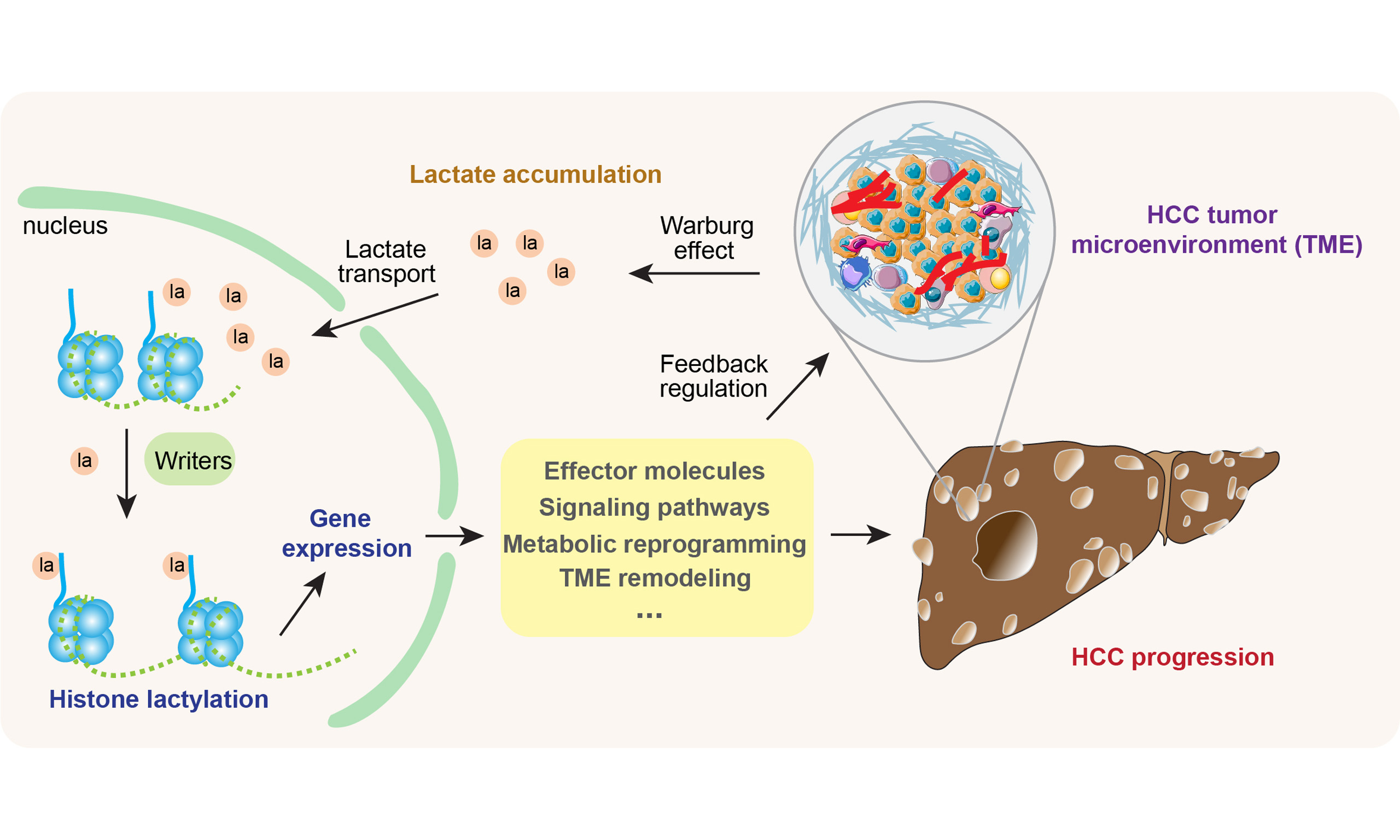
HCC exhibits prominent metabolic reprogramming, characterized by aerobic glycolysis and excessive lactate production[15-17]. Lactate not only acts as an energy substrate or signaling molecule but also directly participates in chromatin modification, imprinting “metabolic memory” on the tumor cell genome. This modification regulates both tumor cell function and the behavior of immune cells in the TME, ultimately shaping a local microenvironment that favors tumor growth and immune escape[8-10,13,18]. Using integrative lactylome and proteome analysis combined with gene targeting tools, recent studies have identified a wide range of novel non-histone proteins that undergo lactylation in HCC, including the metabolic enzyme adenylate kinase 2 (AK2), the transcriptional regulator yes-associated protein (YAP), and the tumor suppressor p53[9,19-22]. Evidence has shown that such non-histone lactylation events, together with histone lactylation, are emerging as key regulatory mechanisms in HCC, governing critical cellular processes including tumor cell proliferation, apoptosis and signal transduction in the TME. Here, we focus on the roles of histone lactylation in HCC.
Mounting evidence suggests that histone lactylation regulates HCC initiation and progression through multiple pathways[2,13,23,24]. This review begins with the molecular characteristics of histone lactylation and its involvement in the HCC immune microenvironment, then details how histone lactylation regulates HCC metabolic reprogramming, associated signaling pathways, and its functional impacts on TME immune cells. Finally, we discuss future research directions and drug development targeting histone lactylation in HCC.
HISTONE LACTYLATION MODIFICATION
Lactate production is a prerequisite for histone lactylation. Owing to the Warburg effect, glucose is metabolized to pyruvate through anaerobic glycolysis; pyruvate is then converted to lactate by lactate dehydrogenase A (LDHA). Lactate is secreted extracellularly via monocarboxylate transporters (MCTs, e.g., MCT4), while intracellular lactate combines with coenzyme A (CoA) under the action of lactate-CoA synthetase to form lactate-CoA-the direct donor for histone lactylation - which provides acyl groups for lysine residue modification. In high-lactate environments, histone lactylation levels increase significantly, indicating it is an epigenetic modification that rapidly responds to cellular metabolic changes[8,9,12].
In 2019, researchers first reported lactate as a substrate for histone modification[11]. Lactate generates a high-energy intermediate, lactate-CoA, under the catalysis of lactate-CoA synthetase (not LDH), and then the lactyl group is transferred to the ε-amino group of histone lysine residues by HATs [e.g., p300/cAMP-response element binding protein(CREB)-binding protein (p300/CBP)] or other lactyltransferases, forming lactylation[7,12,22].
Structurally, histone lactylation shares similarities with traditional histone acetylation (both are acylation modifications) but differs in side-chain structure (lactylation contains a hydroxyl group). Similar to acetylation, lactylation neutralizes the positive charge of lysine, reducing histone-DNA affinity, relaxing chromatin structure, and promoting gene transcription[12,13]. In addition to the classic modification site H3K18la, other known lactylation sites identified so far include H2AK11la, H2AK13la, H2AK115la, H2BK5la, H2BK11la, H2BK15la, H2BK16la, H2BK20la, H2BK23la, H2BK43la, H2BK58la, H2BK85la, H2BK108la, H3K9la, H3K14la, H3K23la, H3K27la, H3K56la, H3K79la and H4K5la, H4K8la, H4K12la, and H4K16la, H4K31la, H4K77la and H4K91la[11,14,23-29]. Among them, H3K18la, H3K23la, and H4K12la have attracted the most attention due to their critical roles in physiological and pathological processes [Figure 1]. Notably, lactylation sites in H2A require further validation, as discrepancies exist between putative H2A sequences and mass spectrometry results[11]. Currently identified “writers” for histone lactylation include acetyltransferase p300/CBP, GCN5 (also known as lysine acetyltransferase 2A, KAT2A), HAT binding to ORC1 (HBO1; also known as KAT7), KAT5 and KAT8. Among them, p300/CBP was the first to be identified by Zhang et al.[11,14,30-35]. Some HDACs [e.g., Sirtuin 1-3 (SIRT1-3), HDAC1-3] act as “erasers” due to their histone delactylase activity[7,12,14] [Figure 1]. Additionally, intracellular lactate concentration strictly regulates histone lactylation levels.
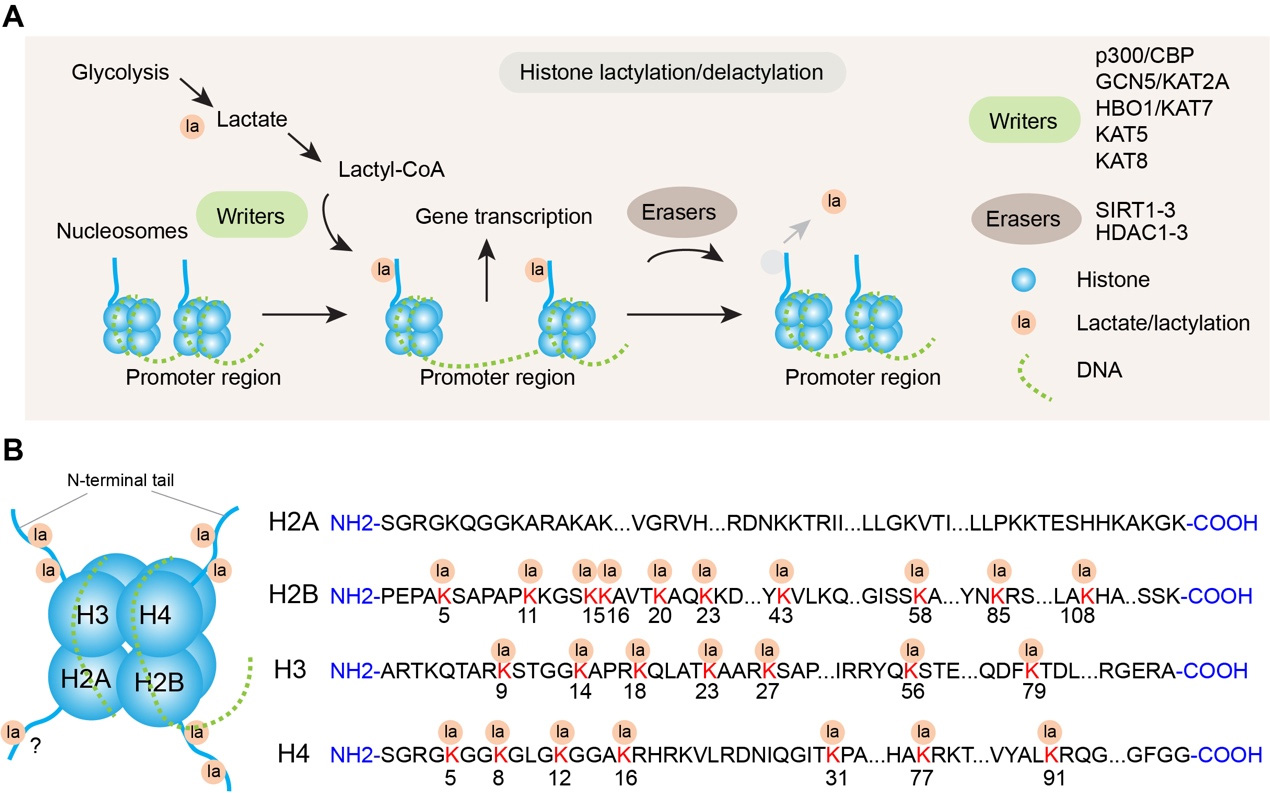
Figure 1. Histone lactylation as a metabolic-epigenetic regulator. (A) Schematic of histone lactylation-mediated gene transcription regulation. Glycolysis generates lactate, which is converted to lactyl-CoA, a key substrate for histone lactylation. “Writers” (e.g., p300/CBP, GCN5/KAT2A) catalyze the addition of lactyl groups (la) to histones in the promoter region of nucleosomes, leading to the activation of gene transcription. Conversely, “Erasers” (e.g., SIRT1-3, HDAC1-3) mediate histone delactylation, removing lactyl modifications and reversing transcriptional activation; (B) Identified lactylation sites on histone N-terminal tails. Lactyl groups (la) are enriched on the N-terminal tails of core histones (H2A, H2B, H3, H4). The sequence of each histone is shown, with numbered positions (e.g., H2BK5, H3K9) indicating confirmed lactylation sites. Note: modification sites in H2A have not been identified in the canonical H2A sequence, despite a previous report[11].
HCC AND TUMOR IMMUNE MICROENVIRONMENT
Etiology of HCC
HCC initiation is closely linked to multiple risk factors, including chronic hepatitis B virus (HBV) infection, chronic hepatitis C virus (HCV) infection, alcoholic liver disease, non-alcoholic fatty liver disease (NAFLD), and aflatoxin exposure[36]. These factors induce long-term liver damage and inflammation, leading to hepatocyte degeneration, necrosis, and proliferation, and ultimately progressing to liver cirrhosis and HCC[15,37-39]. HCC has an insidious onset with no obvious early symptoms; most patients are diagnosed at an advanced stage, missing the optimal window for surgical resection. Current HCC treatments include surgical resection, liver transplantation, interventional therapy, targeted therapy, and immunotherapy, but the overall efficacy remains unsatisfactory, with a low 5-year survival rate[15,37]. Thus, in-depth exploration of HCC pathogenesis and identification of novel diagnostic markers and therapeutic targets are crucial for improving patient prognosis.
HCC immune microenvironment
The tumor immune microenvironment (TIME) refers to a complex microenvironment in tumor tissue composed of immune cells, stromal cells, vascular endothelial cells, cytokines, chemokines, and metabolites. The HCC TIME is highly heterogeneous and complex, with its composition and functional state directly influencing tumor initiation, progression, and treatment response[37]. Immune cells in the HCC TIME primarily include T lymphocytes, B lymphocytes, natural killer (NK) cells, tumor-associated macrophages (TAMs), dendritic cells (DCs), myeloid-derived suppressor cells (MDSCs), cancer-associated fibroblasts (CAFs), and regulatory T cells (Tregs)[38]. Among these, CD8+ cytotoxic T lymphocytes (CTLs) are the main effector cells mediating anti-tumor immunity, which specifically recognize and kill tumor cells; Tregs and MDSCs are key immunosuppressive cells, inhibiting CTL activity through secreted factors [e.g., IL-10, transforming growth factor-β (TGF-β)] to promote immune escape[17,40]; TAMs exert dual roles: M1-type TAMs have anti-tumor activity, while M2-type TAMs promote tumor angiogenesis, invasion, metastasis, and immunosuppression[37,40]. The HCC TIME also exhibits significant metabolic alterations as the Warburg effect increases lactate concentration and decreases pH in the TME. This acidic environment not only directly inhibits immune cell function but also regulates immune cell polarization and activity via epigenetic modifications, further promoting tumor immune escape[41].
MECHANISMS OF HISTONE LACTYLATION-MEDIATED REGULATION IN HCC PROGRESSION
Oncogenic gene activation
Histone lactylation levels are increased in HCC tissues and cells[23,42-44]. This modification is closely associated with the transcriptional activation of pro-tumorigenic genes, which affect cell signal transduction, proliferation, and TME immune escape in HCC [Table 1]. For example, lactylation at H3K18 promotes SRY-box transcription factor 9 (SOX9) expression, accelerating liver fibrosis and HCC initiation[24]. Endothelial cell-specific molecule 1 (ESM1), regulated by histone lactylation, enhances HCC cell proliferation and invasion when highly expressed[23]. Histone lactylation upregulates minichromosome maintenance complex component 7 (MCM7) expression, boosting the transcription of cancer stem cell (CSC)-related genes involved in stemness maintenance and cell proliferation, thereby promoting CSC properties and radioresistance in HCC[43]. Y-Box Binding Protein 1 (YBX1), highly expressed in HCC, regulates glycolytic pathways: enhanced YBX1 and downstream glycolysis-related gene transcription stimulate lactate production, while simultaneously activating p300 transcription (p300 promotes H3K18la at the YBX1 gene promoter). This forms a YBX1/glycolysis/H3K18la positive feedback loop that accelerates HCC progression[45]. However, YBX1-associated signaling pathways in HCC require further investigation. In cisplatin-resistant HCC tissues and cells, H3K18la upregulates the expression of ubiquitin-specific protease 34 (USP34), a key mediator of tumor drug resistance[46]. Moreover, increased H3K18la at the insulin receptor substrate 1 (IRS1) promoter induces IRS1 expression and downstream activation of the phosphatidylinositol 3-kinase/AKT serine/threonine kinase/mechanistic target of rapamycin kinase (PI3K/AKT/mTOR) and mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK) pathways, promoting HCC progression[47]. Collectively, histone lactylation may directly drive HCC progression by regulating downstream target genes involved in tumorigenesis and metastasis [Figure 2], although broader and deeper investigations are still needed.
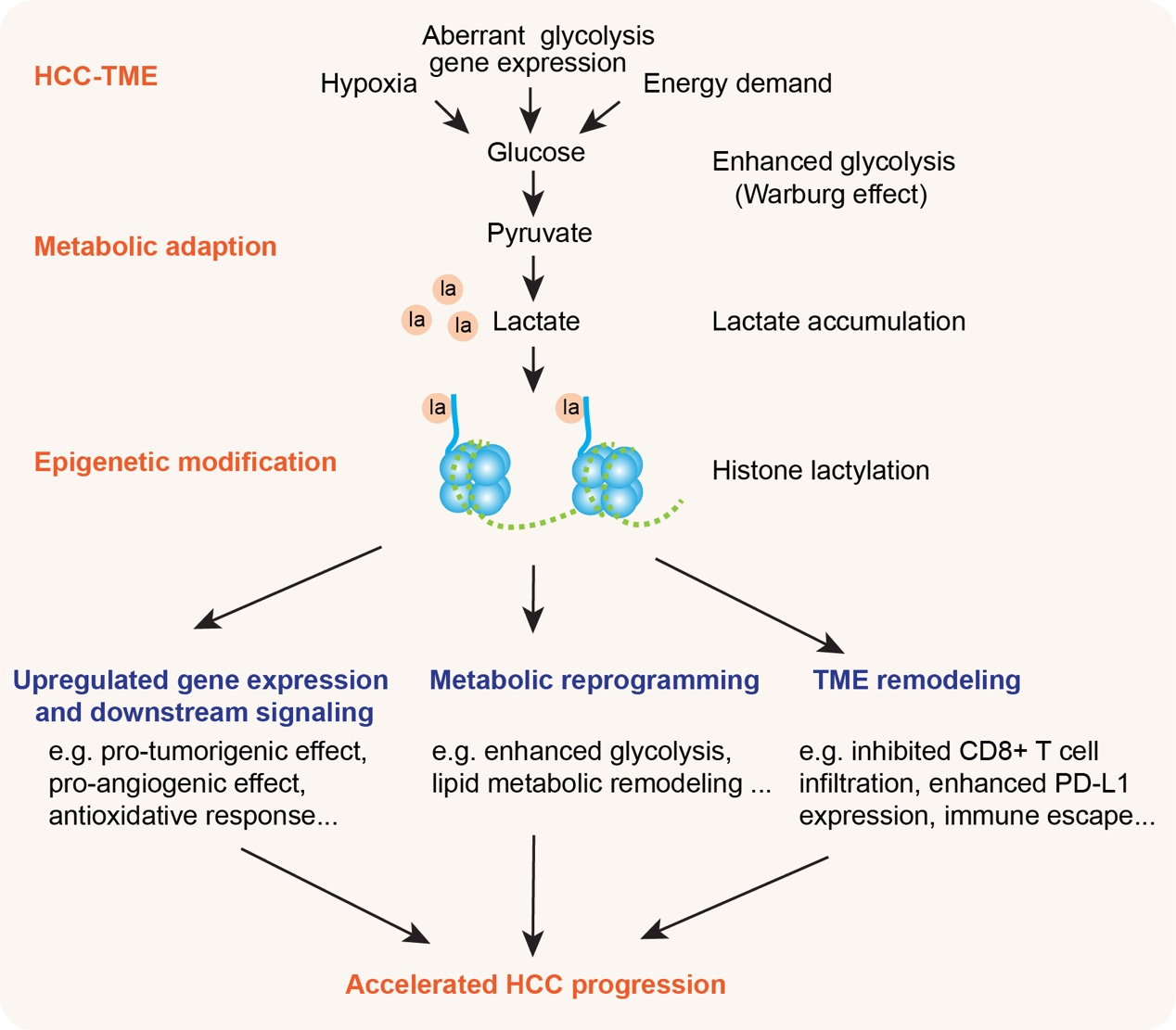
Figure 2. Histone lactylation links metabolic adaptation to HCC progression in the tumor microenvironment (TME). In the HCC-TME, contextual cues (hypoxia, aberrant glycolysis gene expression, high energy demand) drive enhanced glycolysis (the Warburg effect): glucose is metabolized to pyruvate, which is preferentially converted to pyruvate, leading to lactate accumulation. This metabolic adaptation enables epigenetic modification via histone lactylation, which triggers key pro-tumor processes that converge to accelerate HCC progression, establishing a feedforward loop between metabolic adaptation and epigenetic regulation in the HCC-TME. HCC: Hepatocellular carcinoma.
Summary of mechanistic studies on histone lactylation in HCC
| Histone lactylation site | Upstream regulator | Downstream target | Related effectors or pathways | Effects on HCC progression | Reference |
| H3K18la | LDHA upregulation | SOX9 | Not mentioned | Promoted HSC proliferation and accelerated liver fibrosis | [24] |
| H3K9la, H3K56la | Not mentioned | ESM1 | Not mentioned | Enhanced HCC cell proliferation, migration, invasion, and EMT | [23] |
| Pan-Kla | Not mentioned | MCM7 | Not mentioned | Enhanced CSC properties, accelerated CSC proliferation and radio-resistance | [43] |
| H3K18la | YBX1-upregulated glycolysis and p300 activation | YBX1 | Glycolysis pathways | Upregulated glycolysis, lactylation, and HCC progression | [45] |
| H3K18la | LDHA and LDHB upregulation | USP34 | Not mentioned | Upregulated cell proliferation and cisplatin resistance in HCC | [46] |
| H3K18la | PYCR1 upregulation | IRS1 | PI3K/AKT/mTOR and MAPK/ERK pathways | Promoted liver cancer cell proliferation and metastasis | [47] |
| H3K14la | Not mentioned | NEDD4 | NEDD4/PTEN pathway | Exacerbated HCC malignancy and chemotherapy resistance | [48] |
| H3K18la | Not mentioned | HECTD2 | KEAP1/NRF2 pathway | Elevated antioxidative response and subsequent lenvatinib resistance | [49] |
| H3K18la | c-Myc | GP73 | JAK2/STAT3/GRP78 signaling pathway | Enhanced endoplasmic reticulum stress and pro-angiogenic effects | [50] |
| H3K18la | Not mentioned | NFS1 | Ferroptosis pathways | Reduced susceptibility of HCC to ferroptosis and enhanced HCC metastasis | [25] |
Regulation of signaling pathways
At present, multiple classic signaling pathways (e.g., PI3K/Akt/mTOR, Wnt/β-catenin, NF-κB, hypoxia-inducible factor 1α (HIF-1α), and JAK/STAT signaling pathways) have been found to be modulated by histone lactylation in various tumors (see review[13,14,30]). However, evidence for histone lactylation modulating these pathways in HCC remains limited [Figure 2]. Key findings to date are summarized below [Table 1]. Enhanced glycolytic activity has been consistently identified in drug-resistant HCC cells and inhibiting glycolysis effectively suppressed their malignant behavior. Active glycolysis induced lactate accumulation and H3K14la elevation, which upregulate the downstream expression of the E3 ubiquitin ligase Neural precursor cell expressed developmentally down-regulated 4 (NEDD4). NEDD4 mediates ubiquitination and degradation of the tumor suppressor protein phosphatase and tensin homolog (PTEN), exacerbating HCC malignancy and chemotherapy resistance via the H3K14la/NEDD4/PTEN pathway[48]. Similarly, H3K18 lactylation promotes the transcription of another E3 ubiquitin ligase, HECT domain E3 ubiquitin protein ligase 2 (HECTD2), which contributes to the degradation of kelch like ECH associated protein 1 (KEAP1) protein and activation of the anti-oxidative KEAP1/nuclear factor-erythroid 2-related factor 2 (NRF2) signaling pathway[49]. Single-cell spatial transcriptomic landscape of the vasculogenic etiology of HCC has uncovered that Golgi phosphoprotein 73 (GP73)-overexpressing HCC cells exhibit high pro-angiogenic potential in vascular endothelial cells. Mechanistically, the lactylation “writer” enzyme p300 mediates histone lactylation to stimulate the expression of GP73 (a pivotal hub gene), and GP73 then activates the Janus kinase 2/signal transducer and activator of transcription 3 (JAK2/STAT3) signaling pathway, enhancing downstream activation of glucose-regulated protein 78 (GRP78)-induced endoplasmic reticulum stress (ERS). This histone lactylation-regulated GP73/JAK2/STAT3/GRP78 pathway promotes tumor vascular development in HCC[50]. Pyrroline-5-carboxylate reductase 1 (PYCR1) is significantly upregulated in HCC, with high expression correlating with poor prognosis. Transcriptome sequencing and metabolomics analysis in liver cancer cells showed that PYCR1 increases glycolysis and regulates IRS1 transcriptional activity by modifying H3K18la at the IRS1 gene promoter, thus regulating IRS1-mediated PI3K/AKT/mTOR and MAPK/ERK pathways to promote HCC cell proliferation and metastasis[47]. Moreover, in sublethal heat treatment-induced HCC metastasis, integrated proteomic and transcriptomic data with H3K18la-specific chromatin immunoprecipitation (ChIP) revealed that increased H3K18la enhances transcriptional activity of cysteine desulfurase (NFS1, a key enzyme in iron-sulfur cluster biosynthesis), reducing HCC susceptibility to ferroptosis[25]. This evidence suggests that NFS1-mediated ferroptosis signaling is also involved and possibly regulated by histone lactylation in HCC.
Metabolic reprogramming
Metabolic reprogramming is a core hallmark of tumor cells. As a key mediator of this process, lactate promotes continuous glycolysis in tumor cells, enhancing drug resistance and adaptability, thereby driving cancer initiation and progression [Figure 2][10,22,41]. The overall protein lactylation is significantly elevated in HCC tissues, with high lactylation levels correlating with more active glycolysis and a more malignant phenotype[20,51,52]. LDHA, critical for histone lactylation, is highly expressed in HCC cells[21], and high LDHA levels increase intracellular lactate concentration, further promoting histone lactylation[13,21,41]. ChIP sequencing (ChIP-seq) analysis revealed that lactylation modifications are enriched in the promoter regions of genes related to tumor proliferation, metabolic adaptation, hypoxia response, and immune regulation[12,14,22]. This indicates that it is not a random event but selectively reshapes the transcriptome of HCC cells to adapt to their survival needs. Nonetheless, aberrant lactate metabolism is not unique to HCC but a common metabolic hallmark shared by most solid tumors. However, as a central organ for systemic lactate clearance and gluconeogenesis, the liver undergoes a pathological switch during hepatocarcinogenesis, in which hepatocytes transform from lactate consumers to lactate producers. This functional reversal may render the dysregulated lactate metabolism in HCC tissue-specific and disease-specific, providing a unique pathological basis for the metabolism-derived epigenetic response.
Lactate accumulation in HCC increases histone lactylation, which in turn upregulates metabolism-related genes, thus forming a positive feedback loop that reinforces metabolic reprogramming. For example, O-linked N-acetylglucosamine (O-GlcNAc) modification stabilizes YBX1, increasing its expression and nuclear translocation in HCC. Consequently, both the transcription of downstream glycolysis-related genes and lactate production are stimulated. In addition, YBX1 and serine- and arginine-rich splicing factor 10 (SRSF10) upregulate key glycolytic enzymes, leading to lactate accumulation that drives the histone lactylation (particularly H3K18la), which in turn positively regulates YBX1 and SRSF10 gene transcription, establishing a YBX1/SRSF10-glycolysis-H3K18la positive feedback loop that accelerates HCC progression[45,53]. In line with this study, in metabolic dysfunction-associated steatotic liver disease (MASLD), H3K18la binds to the promoters of glycolytic key enzymes [e.g., hexokinase 2 (HK2)], upregulating HK2 expression, enhancing anaerobic glycolysis, and promoting lactate-mediated histone lactylation. This HK2/glycolysis/H3K18la loop ultimately exacerbates metabolic dysregulation and histone lactylation[54].
Lipid metabolism reprogramming also drives HCC initiation and progression. Elevated histone lactylation in HCC induces upregulated expression of the N6-methyladenosine (m6A) reader YTH N6-Methyladenosine RNA Binding Protein C1 (YTHDC1), thereby increasing the stability of m6A-modified long noncoding RNA nuclear paraspeckle assembly transcript 1 (NEAT1). NEAT1 then recruits the HAT p300 to the promoter of the gene encoding stearoyl-CoA desaturase (SCD), activating SCD-associated lipid metabolic processes that accelerate HCC progression[55]. These findings suggest that histone lactylation mediates lipid metabolic remodeling to promote HCC progression.
TME remodeling and immunotherapy resistance
HCC is characterized by a “cold” tumor and an immunosuppressive TME, which limits the efficacy of immune checkpoint blockade therapy based on immune checkpoint inhibitors (ICIs)[37-39]. This “cold” state arises from complex interactions between TME immune cells and their signaling crosstalk[37,41]. While the role of histone lactylation in regulating the TIME has been studied in various tumors (see review[2,28,30,56]), research in HCC has focused primarily on a subset of immune cells within the TME (e.g., CD8+ T cells).
Elevated lactylation correlates with poor prognosis and immunotherapy resistance in HCC [Figure 2]. Inhibition of lactylation enhances CD8+ T-cell infiltration and cytokine production[52,53]. Key findings to date are summarized below. In the HCC TIME, YAP1 upregulates lactate transport and accumulation, increasing p300/CBP catalytic activity and enhancing HDAC1-3 deacetylation - thus stimulating histone lactylation and regulating gene expression. These effects are reversed by dihydroartemisinin (DHA)[44]. Importantly, DHA converts the HCC-TIME from “cold” to “hot” by increasing the abundance and proportion of T cells, NK cells, M1-like TAMs, and DCs (in a CD8+ CTL-dependent manner), enhancing anti-HCC immunity and sensitizing tumors to anti-programmed cell death 1 (PD-1) therapy[44]. As mentioned earlier, SRSF10 positively regulated lactate production, forming an SRSF10/glycolysis/H3K18la positive feedback loop in HCC cells. Lactate accumulation and H3K18la-regulated gene transcription promote M2-type TAM polarization, inhibiting CD8+ T cell enrichment, reducing interferon-γ+ CD8+ T cell proportions, thus creating an immunosuppressive TME. Pharmacological inhibition of SRSF10 improves anti-PD-1 efficacy in murine and human preclinical models[53].
Apart from the mechanisms that histone lactylation induces altered proportions of specific immune cell types in TIME, evidence also shows that this modification regulates the efficacy of immunotherapy by directly tuning programmed cell death 1 ligand 1 (PD-L1) expression at the transcriptional level or via protein stability[52,57]. Protein arginine methyltransferase 3 (PRMT3) interacts with pyruvate dehydrogenase kinase 1 (PDHK1), a key enzyme in glycolysis, and increases its kinase activity, stimulating glycolysis and lactate accumulation in HCC. ChIP assay showed that PRMT3 enhances H3K18la binding to the PD-L1 promoter and upregulates lactate-induced PD-L1 expression. As a consequence, anti-PD-L1 treatment reverses PRMT3-induced tumor growth and restores CD8+ T cell infiltration, highlighting a PRMT3-PDHK1-lactate-PD-L1 axis linking metabolic reprogramming and immune escape[57]. Major vault protein (MVP) is a lactylation-regulated factor. High expression of MVP correlates with resistance to ICI therapy. Histone lactylation-induced MVP upregulation stabilizes PD-L1 by preventing β-transducin repeat-containing protein (β-TrCP)-mediated PD-L1 ubiquitination and subsequent proteasomal degradation, suppressing CD8+ T cell-mediated antitumor immunity. Consistently, pharmacological inhibition of lactylation sensitizes anti-PD-1/PD-L1 therapy in HCC mouse models[52].
In addition to PD-L1, B7 homolog 3 protein (B7-H3, also known as CD276), a newly identified immune checkpoint ligand and a tumor-associated antigen enriched in the TME, has been shown to be regulated by histone lactylation, making it a promising target for cancer immunotherapy[58]. In HCC tissues, B7-H3 expression is linked to glycolysis. Lactate-upregulated H3K18la directly binds to the B7-H3 promoter and increases B7-H3 expression, leading to compromised proportion and cytotoxicity of infiltrating CD8+ T cells. Targeted inhibition of glycolysis and B7-H3 expression enhances the efficacy of anti-PD-1 treatment[59]. In conclusion, targeting histone lactylation in combination with PD-1/PD-L1 blockade offers a promising strategy to overcome immunotherapy resistance in HCC.
Targeting histone lactylation in HCC
Given the critical role of histone lactylation in HCC initiation, progression, and immune escape, targeting this modification has become a novel strategy.
First, inhibition of the production and transport of lactate can downregulate the donor of histone lactylation; therefore, relevant inhibitors are widely used in mechanistic cancer studies including HCC. Pyruvate dehydrogenase kinase 1 (PDK1) inhibitors (e.g., sodium dichloroacetate) and LDH inhibitors (e.g., sodium oxamate) were used to inhibit lactate production by modulating the activities of pyruvate dehydrogenase and lactate dehydrogenase, respectively[11]. Other LDHA inhibitors (e.g., GNE-140, GSK2837808A), metabolic inhibitors (such as the glycolysis inhibitor 2-deoxy-D-glucose), or MCT inhibitors (e.g., AZD3965) are potent modulators to reduce lactate accumulation and histone lactylation[11,12,30]. HK2 and pyruvate kinase M2 isoform (PKM2) in the glycolytic pathway are also potential targets for HCC via histone lactylation, and HK2 inhibitor attenuates hepatic stellate cell (HSC) activation and liver fibrosis (a disease stage before HCC) via histone lactylation[60]. In addition, rotenone, an inhibitor of the mitochondrial respiratory chain complex I that drives cells towards glycolysis, has also been widely used to attenuate glycolysis-induced lactylation[11].
Second, enzymes engaged in histone lactylation modification are promising targets. Although histone lactylation “writers” (e.g., p300/CBP, GCN5/KAT2A, HBO1/KAT7, KAT5, and KAT8) and “erasers” (e.g., HDAC1-3 and SIRT1-3) have been identified, p300 is the most well-studied and the role of other non-histone “writers” and “erasers” in HCC remains unclear[12,14,30,31,33-35]. The p300 inhibitor C646 may inhibit histone lactylation, suppressing HCC cell proliferation and immunity[12]; while activating delactylases (such as SIRT1/2 agonists, including resveratrol) may promote the removal of lactylation, reducing histone lactylation levels[7,12]. Agonists and inhibitors for other modification enzymes require further investigation in HCC. It is worth noting that current research on the detection of histone lactylation is heavily reliant on site-specific antibodies such as anti-H3K18la, yet the specificity and cross-reactivity of these antibodies remain to be widely validated. In addition, unlike mature high-throughput quantitative detection technologies for post-translational modifications (PTMs) such as phosphoproteomics, there is a lack of standardized, high-sensitivity, and high-throughput quantification methods for histone lactylation sites at present. The advent of such technologies will not only broaden the systematic identification and quantitative analysis of lactylation modification sites in HCC cells and immune microenvironment cells, but also facilitate the in-depth exploration of the dynamic changes and functional differences of lactylation at different sites during HCC progression.
Third, targeting the effector proteins of histone lactylation modification may block downstream signaling pathways in HCC. A body of studies has shown that inhibition of PD-L1 expression enhances the immunogenicity of tumor cells and improves ICI efficacy[59]. Therefore, combining metabolic/epigenetic interventions with immunotherapies [e.g., anti-PD-1/PD-L1, anti-cytotoxic T-lymphocyte-associated protein 4 (CTLA-4)] is currently the most promising clinical translation path. Notably, although elevated lactate secretion in HCC promotes Treg cell accumulation and ICI resistance[61], the role of histone lactylation in anti-CTLA-4 therapy for HCC has not been reported yet. Therefore, targeting histone lactylation may serve as a key combination therapy strategy to improve the HCC immunotherapy efficacy.
Clinical translation
Although targeting general lactate metabolism related to histone lactylation has emerged as a promising therapeutic strategy for HCC, including the use of LDHA inhibitors, MCT blockers, and glycolysis inhibitors, the clinical translation of these strategies is still faced with multiple challenges and potential risks that cannot be ignored. First, lactate is an important physiological metabolite in the body, which also plays a key role in energy supply and metabolic regulation of normal tissues such as skeletal muscle and brain; non-specific inhibition of lactate metabolism may disrupt the normal physiological functions of these tissues and cause severe off-target toxic side effects. Second, most of the small-molecule inhibitors targeting lactate metabolism have poor selectivity, which are difficult to specifically act on tumor cells and easily affect the metabolic balance of normal cells. Third, lactate in the HCC-TIME has complex and diverse sources, which are not only secreted by HCC cells with abnormal glycolysis, but also largely produced by stromal cells such as CAFs and TAMs in the microenvironment; single-target inhibition of lactate production in tumor cells may be difficult to completely eliminate the high-lactate microenvironment. These problems need to be solved through the development of cell-specific targeted delivery systems and multi-target combined intervention strategies in subsequent research. Moreover, precise cell/site-specific lactylation modulation remains unachievable, in vivo and clinical evidence for lactylation’s pathogenic and therapeutic roles is scarce, and lactylation’s complex crosstalk with other epigenetic and metabolic regulatory networks is poorly understood. All these factors are essential for the accurate functional characterization and translational development of lactylation-based therapies.
Specifically, drug development programs targeting histone lactylation remain in the early stages of preclinical exploration, and to date, no such candidates have advanced to clinical trial evaluation. Major challenges ahead include: (a) the identification of histone lactylation modification enzymes is not yet complete, leading to a lack of specific regulatory targets; (b) site-specific histone lactylation may exert distinct biological functions, making site-specific regulation a key technical barrier; (c) drugs targeting histone lactylation may disrupt the lactate metabolism and histone modification of normal cells, causing potential toxic side effects. Thus, the precise balance of efficacy and safety needs to be guaranteed.
From a clinical perspective, relevant applications are in urgent need: (1) developing specific histone lactylation detection tools (e.g., antibodies that recognize specific histone lactylation sites and high-throughput quantitative detection technologies) and regulatory drugs (e.g., enzyme inhibitors/agonists) to lay the foundation for clinical applications; (2) exploring combination therapies (e.g., histone lactylation targeting combined with ICIs, chemotherapy, or radiotherapy) to enhance HCC treatment efficacy; (3) conducting clinical translational research by detecting histone lactylation levels in HCC patient tumors to evaluate their potential as a diagnostic/prognostic marker, and initiating clinical trials of lactylation-targeting drugs to verify safety and efficacy.
CONCLUSION AND PERSPECTIVES
While research on histone lactylation modification in HCC has achieved significant advances, key mechanisms remain to be further studied. For example, our understanding of the roles of lactylation enzymes (writers and erasers) and site-specific histone lactylation is critical for deciphering HCC pathogenesis and TIME regulation. Additionally, research on downstream signaling pathways and the crosstalk between histone lactylation and other histone modifications is limited. With advances in high-throughput technologies (e.g., spatial/spatiotemporal transcriptomics and single-cell ChIP-seq), it will be possible to construct a cell-specific histone lactylation map of HCC and dissect regulatory mechanisms across different cell types and progression stages. Furthermore, investigating the role of histone lactylation in HCC of distinct etiologies (e.g., HBV/HCV infection, NAFLD) is essential for developing personalized therapies.
In summary, histone lactylation plays a key regulatory role in the HCC immune microenvironment, involving metabolic reprogramming, signaling pathway regulation, and immune cell function regulation. In-depth study of these mechanisms will not only expand our understanding of the HCC pathogenesis but also provide a theoretical basis for the development of novel diagnostic markers and therapeutic strategies. In the future, integrating multi-omics and advanced imaging/genetic technologies, and rational drug design will enable targeting of histone lactylation in the metabolic-epigenetic axis, offering more efficient treatments for HCC patients.
DECLARATIONS
Authors’ contributions
Designed the structure of the review, edited and revised the manuscript: Li C, Wu L
Collected data and drafted the manuscript: Fu C, Li C
Drafted the figures: Fu C, Wu L
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
During the preparation of this manuscript, the AI tool Doubao (online webpage version, updated in real time) was used solely for language editing. The tool did not influence the study design, data collection, analysis, interpretation, or the scientific content of the work. All authors take full responsibility for the accuracy, integrity, and final content of the manuscript.
Financial support and sponsorship
This study was supported by the Science and Technology Project of Jiangxi Traditional Chinese Medicine Administration to Wu L (2023B1242) and the National Natural Science Foundation of China (31900692, 32070961), Hubei Natural Science Foundation project (2024AFB558) to Li C.
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Webster AK, Phillips PC. Epigenetics and individuality: from concepts to causality across timescales. Nat Rev Genet. 2025;26:406-23.
2. Easwaran H, Weeraratna AT. Unravelling the genetics and epigenetics of the ageing tumour microenvironment in cancer. Nat Rev Cancer. 2025;25:828-47.
3. Mehler M. Epigenetic principles and mechanisms underlying nervous system functions in health and disease. Prog Neurobiol. 2008;86:305-41.
5. Lennartsson A, Ekwall K. Histone modification patterns and epigenetic codes. Biochim Biophys Acta Gen Subj. 2009;1790:863-8.
6. Villar-garea A, Imhof A. The analysis of histone modifications. Biochim Biophys Acta Proteins Proteom. 2006;1764:1932-9.
7. Chen J, Huang Z, Chen Y, et al. Lactate and lactylation in cancer. Sig Transduct Target Ther. 2025;10:38.
8. Yang B, Li L, Shi D, Zhong T, Xiong H. Lactylation and antitumor immunity. Front Immunol. 2025;16:1690068.
9. Zhu W, Fan C, Hou Y, Zhang Y. Lactylation in tumor microenvironment and immunotherapy resistance: new mechanisms and challenges. Cancer Lett. 2025;627:217835.
10. Feng X, Li D, Wang P, Li X, Li G. Lactylation in cancer: unlocking the key to drug resistance and therapeutic breakthroughs. Oncol Res. 2025;33:3327-46.
11. Zhang D, Tang Z, Huang H, et al. Metabolic regulation of gene expression by histone lactylation. Nature. 2019;574:575-80.
12. Xiao S, Zhang S, Sun K, Huang Q, Li Q, Hu C. Lactate and lactylation: molecular insights into histone and non-histone lactylation in tumor progression, tumor immune microenvironment, and therapeutic strategies. Biomark Res. 2025;13:134.
13. Yang Y, Luo N, Gong Z, Zhou W, Ku Y, Chen Y. Lactate and lysine lactylation of histone regulate transcription in cancer. Heliyon. 2024;10:e38426.
14. Peng X, Du J. Histone and non-histone lactylation: molecular mechanisms, biological functions, diseases, and therapeutic targets. Mol Biomed. 2025;6:38.
15. Barcena-varela M, Monga SP, Lujambio A. Precision models in hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2024;22:191-205.
16. Zhao Z, Cui T, Wei F, et al. Wnt/β-Catenin signaling pathway in hepatocellular carcinoma: pathogenic role and therapeutic target. Front Oncol. 2024;14:1367364.
17. Yang H, Li J, Niu Y, et al. Interactions between the metabolic reprogramming of liver cancer and tumor microenvironment. Front Immunol. 2025;16:1494788.
18. Zhang X, Liang C, Wu C, et al. A rising star involved in tumour immunity: lactylation. J Cell Mol Med. 2024;28:e70146.
19. Zong Z, Xie F, Wang S, et al. Alanyl-tRNA synthetase, AARS1, is a lactate sensor and lactyltransferase that lactylates p53 and contributes to tumorigenesis. Cell. 2024;187:2375-2392.e33.
20. Yang Z, Yan C, Ma J, et al. Lactylome analysis suggests lactylation-dependent mechanisms of metabolic adaptation in hepatocellular carcinoma. Nat Metab. 2023;5:61-79.
21. Wei X, Zou L, Huang Y, et al. LDHA-mediated YAP lactylation promotes the tumor progression of hepatocellular carcinoma by inducing YAP dephosphorylation and activation. Biol Direct. 2025;20:64.
22. Zhao L, Cui H, Li Y, Ye Y, Shen Z. Metabolite to modifier: lactate and lactylation in the evolution of tumors. MedComm. 2025;6:e70413.
23. Zhao P, Qiao C, Wang J, Zhou Y, Zhang C. Histone lactylation facilitates hepatocellular carcinoma progression by upregulating endothelial cell‐specific molecule 1 expression. Mol Carcinog. 2024;63:2078-89.
24. Wu S, Li J, Zhan Y. H3K18 lactylation accelerates liver fibrosis progression through facilitating SOX9 transcription. Exp Cell Res. 2024;440:114135.
25. Huang J, Xie H, Li J, et al. Histone lactylation drives liver cancer metastasis by facilitating NSF1-mediated ferroptosis resistance after microwave ablation. Redox Biology. 2025;81:103553.
26. Li L, Dong J, Xu C, Wang S. Lactate drives senescence-resistant lineages in hepatocellular carcinoma via histone H2B lactylation of NDRG1. Cancer Lett. 2025;616:217567.
27. Xu H, Li L, Wang S, et al. Royal jelly acid suppresses hepatocellular carcinoma tumorigenicity by inhibiting H3 histone lactylation at H3K9la and H3K14la sites. Phytomedicine. 2023;118:154940.
28. Xu K, Zhang K, Wang Y, Gu Y. Comprehensive review of histone lactylation: structure, function, and therapeutic targets. Biochem Pharmacol. 2024;225:116331.
29. Li H, Sun L, Gao P, Hu H. Lactylation in cancer: current understanding and challenges. Cancer Cell. 2024;42:1803-7.
30. Jia Z, Lu S, Wang Z, Ge P. From mechanism to targeted therapy: advances in histone lactylationdriven cancer progression (review). Oncol Lett. 2025;31:1-17.
31. Qin Q, Wang D, Qu Y, et al. Enhanced glycolysis-derived lactate promotes microglial activation in Parkinson’s disease via histone lactylation. npj Parkinsons Dis. 2025;11:3.
32. Wang N, Wang W, Wang X, et al. Histone lactylation boosts reparative gene activation post-myocardial infarction. Circ Res. 2022;131:893-908.
33. Zhu R, Ye X, Lu X, et al. ACSS2 acts as a lactyl-CoA synthetase and couples KAT2A to function as a lactyltransferase for histone lactylation and tumor immune evasion. Cell Metab. 2025;37:361-376.e7.
34. Niu Z, Chen C, Wang S, et al. HBO1 catalyzes lysine lactylation and mediates histone H3K9la to regulate gene transcription. Nat Commun. 2024;15:3561.
35. Zou Y, Cao M, Tai M, et al. A feedback loop driven by H4K12 lactylation and HDAC3 in macrophages regulates lactate‐induced collagen synthesis in fibroblasts via the TGF‐β signaling. Adv Sci. 2025;12:2411408.
36. Llovet JM, Kelley RK, Villanueva A, et al. Hepatocellular carcinoma. Nat Rev Dis Primers. 2021;7:6.
37. Luo X, He X, Zhang X, et al. Hepatocellular carcinoma: signaling pathways, targeted therapy, and immunotherapy. MedComm. 2024;5:e474.
38. Wang J, Liu C, Hu R, Wu L, Li C. Statin therapy: a potential adjuvant to immunotherapies in hepatocellular carcinoma. Front Pharmacol. 2024;15:1324140.
39. Zhang J, Hu C, Xie X, Qi L, Li C, Li S. Immune checkpoint inhibitors in HBV-caused hepatocellular carcinoma therapy. Vaccines. 2023;11:614.
40. Cheng K, Cai N, Zhu J, Yang X, Liang H, Zhang W. Tumor‐associated macrophages in liver cancer: from mechanisms to therapy. Cancer Commun. 2022;42:1112-40.
41. Yang Y, Gao Y, Xiong Y, et al. Research progress of warburg effect in hepatocellular carcinoma. Front Biosci. 2024;29:178.
42. Pan L, Feng F, Wu J, et al. Demethylzeylasteral targets lactate by inhibiting histone lactylation to suppress the tumorigenicity of liver cancer stem cells. Pharmacol Res. 2022;181:106270.
43. Liu Z, Han J, Su S, et al. Histone lactylation facilitates MCM7 expression to maintain stemness and radio-resistance in hepatocellular carcinoma. Biochem Pharmacol. 2025;236:116887.
44. Gao Y, Gong Y, Song X, et al. Dihydroartemisinin inhibits histone lactylation through YAP1 to act as a ‘hot’ switch for ‘cold’ tumor in hepatocellular carcinoma. Phytomedicine. 2025;148:157307.
45. Ji Y, Xu Z, Tang L, et al. O-GlcNAcylation of YBX1 drives a glycolysis-histone lactylation feedback loop in hepatocellular carcinoma. Cancer Lett. 2025;631:217957.
46. Fan M, Liu JS, Wei XL, Nie Y, Liu HL. Histone lactylation-driven ubiquitin-specific protease 34 promotes cisplatin resistance in hepatocellular carcinoma. Gastroenterol Res. 2025;18:23-30.
47. Wang H, Xu M, Zhang T, et al. PYCR1 promotes liver cancer cell growth and metastasis by regulating IRS1 expression through lactylation modification. Clin Transl Med.2024;14:e70045.
48. Zeng Y, Jiang H, Chen Z, et al. Histone lactylation promotes multidrug resistance in hepatocellular carcinoma by forming a positive feedback loop with PTEN. Cell Death Dis. 2025;16:59.
49. Dong R, Fei Y, He Y, et al. Lactylation‐driven HECTD2 limits the response of hepatocellular carcinoma to lenvatinib. Adv Sci. 2025;12:2412559.
50. Ye J, Gao X, Huang X, et al. Integrating single-cell and spatial transcriptomics to uncover and elucidate GP73-mediated pro-angiogenic regulatory networks in hepatocellular carcinoma. Research. 2024;7:0387.
51. Hong H, Chen X, Wang H, Gu X, Yuan Y, Zhang Z. Global profiling of protein lysine lactylation and potential target modified protein analysis in hepatocellular carcinoma. Proteomics. 2023;23:2200432.
52. Liu S, Pan Y, Liu W, et al. Lactylation-driven MVP upregulation boosts immunotherapy resistance by inhibiting PD-L1 degradation in hepatocellular carcinoma. J Immunother Cancer. 2025;13:e012230.
53. Cai J, Song L, Zhang F, et al. Targeting SRSF10 might inhibit M2 macrophage polarization and potentiate anti‐PD‐1 therapy in hepatocellular carcinoma. Cancer Commun. 2024;44:1231-60.
54. Li J, Chen X, Song S, et al. Hexokinase 2-mediated metabolic stress and inflammation burden of liver macrophages via histone lactylation in MASLD. Cell Reports. 2025;44:115350.
55. Du W, Tan S, Peng Y, et al. Histone lactylation-driven YTHDC1 promotes hepatocellular carcinoma progression via lipid metabolism remodeling. Cancer Lett. 2025;611:217426.
56. Wang Z, Liu Z, Lv M, Luan Z, Li T, Hu J. Novel histone modifications and liver cancer: emerging frontiers in epigenetic regulation. Clin Epigenet. 2025;17:30.
57. Ding C, Yan F, Xu B, et al. PRMT3 drives PD-L1-mediated immune escape through activating PDHK1-regulated glycolysis in hepatocellular carcinoma. Cell Death Dis. 2025;16:158.
58. Corrigan DT, Tanwar A, Du M, Martin AM, Zang X. The B7-H3 (CD276) pathway: emerging biology and clinical therapeutics. Trends Pharmacol Sci. 2025;46:975-88.
59. Ma Z, Yang J, Jia W, et al. Histone lactylation-driven B7-H3 expression promotes tumor immune evasion. Theranostics. 2025;15:2338-59.
60. Rho H, Terry AR, Chronis C, Hay N. Hexokinase 2-mediated gene expression via histone lactylation is required for hepatic stellate cell activation and liver fibrosis. Cell Metab. 2023;35:1406-1423.e8.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0











Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].