

The clinical landscape of cutaneous neurofibromas in neurofibromatosis type 1
 0
0 
Abstract
Neurofibromatosis type 1 (NF1) is a hereditary tumor predisposition syndrome that predisposes patients to tumors derived from the neural crest cell population. One of the most prominent and well-recognized features is the proclivity for nerve sheath tumors of the skin known as cutaneous neurofibromas (CNs). These tumors are benign and have self-limited growth, but they exert a strong negative impact on patients’ quality of life. The only effective treatments currently are procedural, and there are no available medications. This review addresses the cellular and molecular characteristics of cutaneous neurofibromas with a focus on identifying novel therapeutic targets that could complement existing approaches. Preclinical models, tumor evolution throughout the lifespan, genetic associations with tumor phenotype, and a brief history of interventional clinical trials are also discussed.
Keywords
INTRODUCTION
Neurofibromatosis Type 1 (NF1) is a relatively common autosomal dominant tumor-predilection syndrome and neurocristopathy affecting approximately 1 in 3,000 individuals regardless of sex or ethnicity. Its pathoetiology is dependent on the monoallelic loss of function of NF1, resulting in haploinsufficiency in every tissue of the body. Subsequent somatic loss of the remaining allele results in disease manifestations such as peripheral nerve sheath tumors of the Schwann cell ontology, aberrant bone formation/reabsorption, and low-grade gliomas of the brain, as well as other tumors of neural crest-derived tissues[1,2]. The protein product of NF1, neurofibromin, serves to positively regulate adenylyl cyclase, inactivate rat sarcoma (Ras), inhibit mitosis and induce apoptosis, and inhibit cell adhesion and motility. With germline haploinsufficiency of neurofibromin, Ras signaling escalates, leading to downstream mitogen-activated protein kinase kinase (MEK) activation. Cutaneous neurofibromas (CNs) are benign tumors of the dermis that arise from biallelic loss of NF1 in Schwann cell-like tumor cells and occur in 99% of NF1 patients. They typically begin to develop in late childhood/early adolescence and have self-limited growth trajectories, but lifetime accumulation can ultimately involve the entire integument. CNs can remain small/barely perceptible, or they may grow to large sizes, sometimes reaching many centimeters in diameter. Typically, each patient exhibits a characteristic number and size distribution of tumors, which can range from just a few to tens of thousands. Although individual tumors eventually enter a state of senescence, their ultimate size cannot be precisely predicted.
CNs are primarily associated with physical deformity that can lead to embarrassment, a tendency to cover up the skin with bulky clothing, barriers to intimacy, and socioeconomic disparity due to fewer opportunities for client-facing jobs. CNs can be pruritic or painful and may catch on clothing or jewelry, resulting in bleeding, swelling, and irritation. NF1 quality of life (QOL) metrics have identified that CNs play a major role in the negative impact of NF1 on mood and social interaction[3-5]. The scientific establishment has recognized the importance of understanding CN biology vis-à-vis other tumor types, and of identifying tolerable treatments with satisfactory outcomes for patients[6]. This review article summarizes what is known about CNs, including appearance, tumor initiation, preclinical models, the ultrastructure of CNs, and barriers/challenges of developing further therapies.
CNs HAVE A RANGE OF APPEARANCES
The classical appearance of CNs is that of a skin-colored, mildly erythematous, or rarely hypermelanotic globular lesion with a broad base, typically measuring from a few millimeters to several centimeters in maximal cross-sectional diameter. However, there are many clinical variants [Figure 1]. Tumors may be pedunculated - resembling a ball on a chain (usually more erythematous than the surrounding unaffected skin) - sessile, flat, or globular. Small nascent or latent CNs may only be identifiable using ultrasound imaging. Some CNs present as plaque-like lesions with distinct borders but a more planar appearance, while others are diffuse or dispersed, covering larger anatomic regions and resulting in areas of thickened, irregular skin, with or without the characteristic myxoid or collagenous stroma. Subcutaneous neurofibromas typically have indistinct borders and a more violaceous coloration. On cross-sectional gross pathology, CNs appear as a typically well-delineated white rubbery nodular lesion within the dermis, with an overlying grenz zone of normal papillary dermis, often extending into the subdermis with a barbell-like contraction at the plane of surrounding skin. This can be contrasted with plexiform neurofibromas involving the cutis, which perioperatively are more likely to have a multi-nodular or band-like ultrastructure, and can be more adherent to the overlying skin. Some plexiform neurofibromas involving the skin appear as rugate elephantine hyperpigmented exophytic lesions, whereas others may be confused with a large subcutaneous neurofibroma.
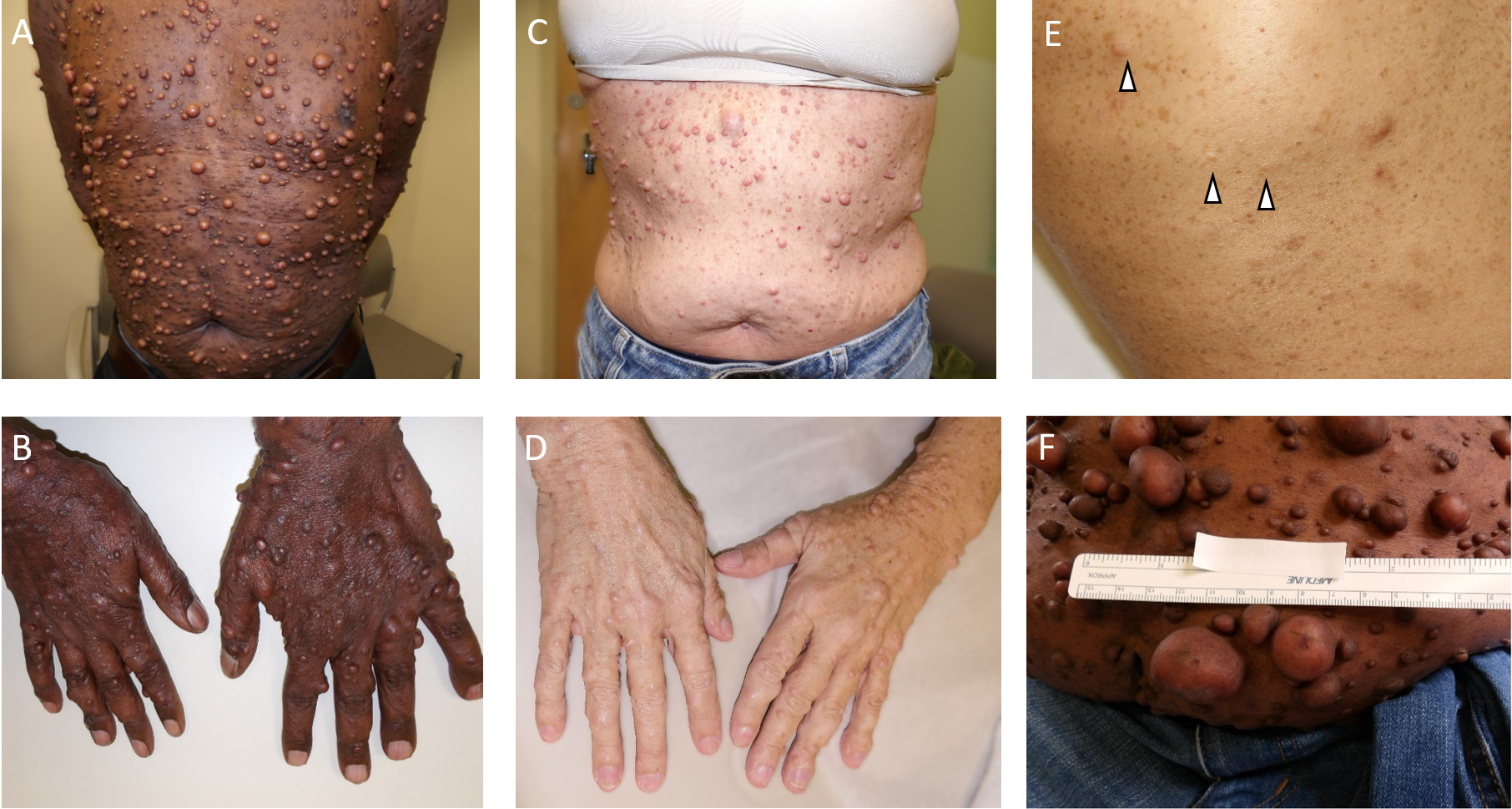
Figure 1. Examples of cutaneous neurofibromas. Moderate to severe burden of average-sized cutaneous neurofibromas on a male with Fitzpatrick Skin Type 6 on (A) chest and abdomen, and (B) hands; (C) and (D) Moderate burden of average-sized cutaneous neurofibromas on the chest and hands, respectively, of a woman with Fitzpatrick Skin Type 1; (E) Mild case of cutaneous neurofibromas (arrow heads) on a background of excess skin freckling; (F) A moderate-severe case of cutaneous neurofibromas on the abdomen. Ruler indicates approximate size of the lesions.
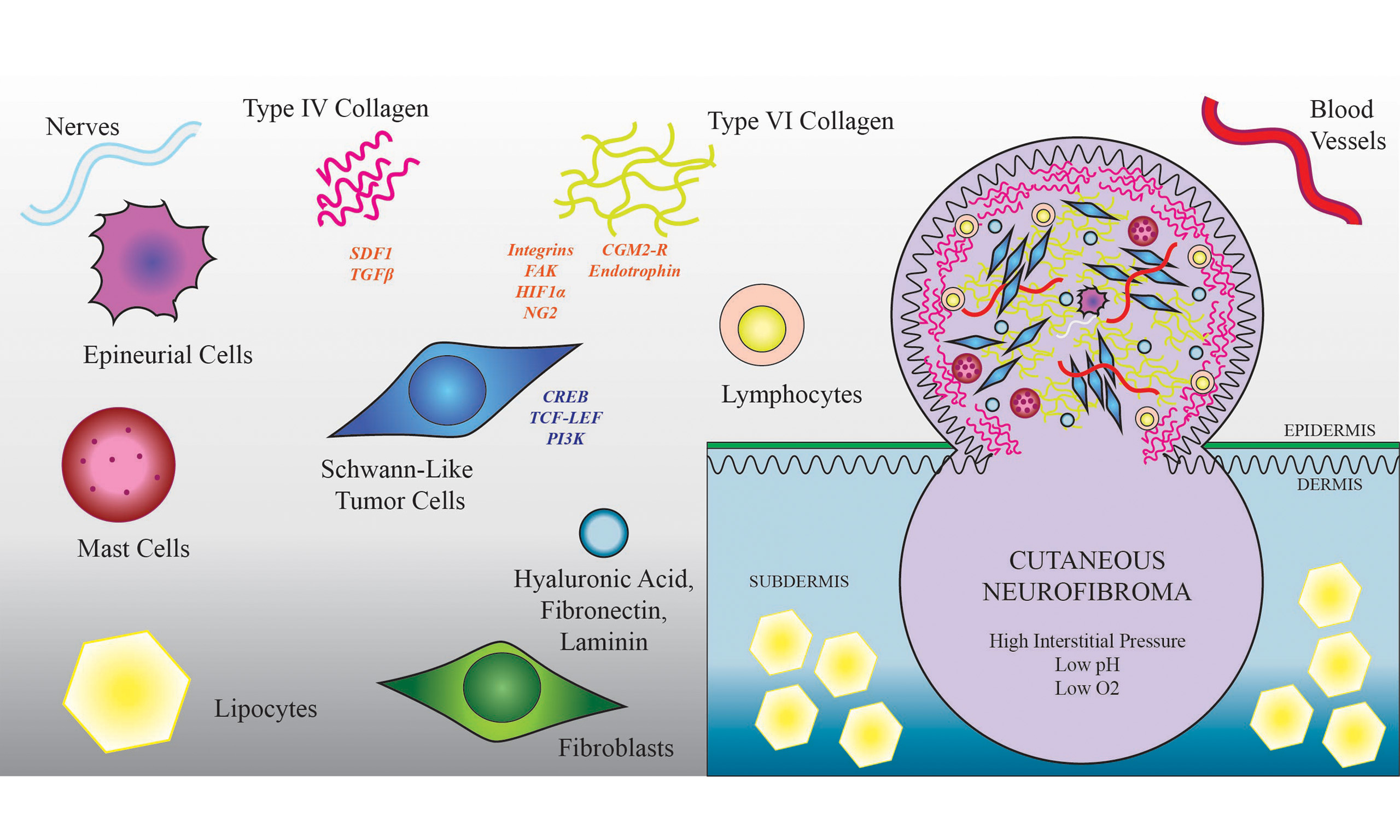
Histopathologically, CNs include abnormal cutaneous nerve endings (myelinated and unmyelinated) and nerve-dissociated bipolar Schwann-like tumor cells lacking an organized basal lamina embedded in a generous supply of extracellular matrix (ECM) with the addition of hyperplastic fibroblasts. CNs can be distinguished from plexiform neurofibromas of the cutis based on the histologic presence of microscopic thickened vermian nerve bundles seen in PNs. However, this definitive feature may not be captured in the sample analyzed by the pathologist; therefore, clinical acumen is important for differentiation. The nerves in CNs include both myelinated and non-myelinated axons. Compared with the surrounding skin, CNs are well-vascularized, containing small arterioles as well as arterial and venous capillaries typically consisting of a single endothelial cell layer with or without subendothelial smooth muscle cells or pericytes, surrounded by a vessel basal lamina and a thickened adventitia. The intima and media layers are also enlarged. On electron microscopy, there are corpuscle-like bodies surrounding small blood vessels or myelinated vessels, composed of lamellar Schwann and perineurial cells with long processes, fine collagen fibers, and basal lamina. A pyramidal cell type with long tripartite processes in connection with morphologically similar cells was identified on electron microscopy as a “covering cell”[7].
These tumors contain a high percentage of infiltrating tumor-supporting cells with atypical proliferative and signaling capabilities associated with haploinsufficiency of neurofibromin from the germline mutation of NF1. Aside from spindle-shaped Schwann-like tumor cells that form the tumor stroma (immunohistochemically identified by Sox10 or S100 staining), there are plentiful fibroblasts (including endoneurial fibroblast-like cells) and perineurial-like cells, as well as endothelial cells and pericytes. There is a robust immune cell component including tumor-associated lymphocytes, dendritic cells, mast cells, and macrophages[8-11]. Occasionally, extensive eosinophils are seen, although this is not characteristic.
Schwann-like cells do not normally proliferate within the dermis, and thus, the identification of tumor stromal cells in a cutaneous biopsy - characterized by fusiform cells irregularly distributed within the ECM on hematoxylin and eosin (H&E) staining or by immunohistochemical (IHC) detection of Schwann cell markers S100 and Sox-10 - is generally accepted as diagnostic of a tumor of the Schwann cell lineage. Beyond this classic neurofibroma histopathologic phenotype, additional more unusual morphologic variants also exist[12]. CNs are difficult to distinguish from cutaneous schwannomas on routine H&E staining, and the need for distinction may not be clinically apparent to the pathologist as both are benign tumors. If the patient does not yet have a genetic diagnosis and/or does not carry the typical features of NF1, two distinct tumors can be submitted for next-generation sequencing of somatic mutation analysis with attention to alterations of chromosome 22q11. If > 1 tumors have different mutations or alterations involving the NF2 gene and no identified alteration of NF1, this would indicate non-NF2-related schwannomatosis rather than NF1. Diagnosticians should carefully consider the possibility of schwannomatosis as a diagnosis for individuals who do not have an identified germline NF1 mutation and who do not fully meet diagnostic requirements for NF1, but who harbor internal nerve sheath tumors and have at least one resected skin lesion identified as a “neurofibroma” based on H&E.
THE EXTRACELLULAR MATRIX INCREASES INTERSTITIAL PRESSURE
The ECM makes up ~50% of the dry weight of the tumor, which is about half of that in skin but twice the concentration found in peripheral nerve endoneurium. The concentration of glycosaminoglycans, particularly hyaluronic acid, is up to 10 times higher in CNs than in the surrounding skin[13]. In CNs, collagen is organized into densely packed, haphazardly splayed bundles, interspersed with intermittent elastin or reticular fibers that surround disorganized, spindle-shaped tumor cells. CNs are physically characterized by high internal tissue tension despite lacking a true capsule. This internal pressure becomes evident during resection at the skin plane, wherein the residual hypodermal tumor often protrudes or “balloons” outward through the incision, driven by the newly created pressure gradient.
CNs are mostly composed of ECM, including fibrillary collagen types (COL) I, II, III, IV, V, network collagen type VI (COLVI) and basement membrane collagen type XV, plasma-derived fibronectin, hyaluronic acid, and Laminin isotype subunit A[14,15]. COLI and COLIII have a filamentous ultrastructure and are involved in fibrosis, and COLIII confers a promitotic cellular phenotype and may contribute to drug resistance[16]. Cultured fibroblasts from CNs primarily produce COLI > COLIII, whereas COLIV is secreted by Schwann-like tumor cells into a basement membrane-like structure coating each cell[13]. In normal skin, COLVI typically resides between the basement membrane and the interstitium. It is produced by Schwann cells to guide remyelination after nerve injury[17,18] and is required for the organization of acetylcholine receptors at the neuromuscular junction[19]. COLVI is also found abundantly in normal skin at the dermal-epidermal border, where it contributes to the near-impenetrable skin barrier to environmental exposures, as well as within skeletal muscle, the cerebral meninges, and in disease states such as glioblastoma and Alzheimer’s disease[20,21].
ECM components are differentially produced by Schwann-derived tumor cells, perineurial cells, and
The collagenolysis of COLVI is not yet fully understood; however, matrix metalloproteases (MMPs) 2, 7, 9, 11, and 14 are able to digest moieties[23]. The major anabolic machinery for collagen resides within the cell cytoplasm. Collagens bind to integrin proteins at the cell membrane and a small peptide moiety is cleaved, which signals
In addition to providing structure and altering diffusion characteristics, ECM contributes to the tumor microenvironment by binding to cell surface proteins to enact diverse pro-tumorigenic signaling pathways within both stromal and tumor-associated NF1 heterozygous cells. COLVI binds to neural/glial antigen 2 (NG2), integrins, or the capillary morphogenesis gene 2 (CMG2/ANTXR2) receptor and activates the cyclic AMP response element-binding protein (CREB)/AKT/β-catenin/T cell factor/lymphoid enhancer factor pathway[24] (TCF-LEF) and phosphatidylinositol-3 kinase (PI3K) signaling[20]. The C-terminal of COLVI (A3) can be proteolytically cleaved, releasing the signaling factor endotrophin, which promotes fibrosis and inflammation. COLVI ultrastructurally interacts with biglycan, fibronectin, decorin, von Willebrand factor (vWF), vWF-A domain-related protein (WARP), fibulin, heparin sulfate, and other collagens, notably COLI and COLIV. COLVI is implicated in the inhibition of apoptosis, the maintenance of a stem-like quality, the promotion of tumor growth, and the activation of autophagy[25].
COLIV is less well-studied than COLVI in tumor biology. It has been proposed as an inducer of
Hyaluronic acid and collagen deposition are associated with high tumor interstitial pressure, low oxygen tension, microvascular collapse, and hypoxia signaling in tumor cells[28], and collagen density correlates negatively with T cell migration into tumors and positively with macrophage-dependent immunosuppression[29]. The overall effect of this dense irregular ECM deposition is to block drug diffusion and immune cell migration, reduce glucose and oxygen supply, increase drug efflux via hypoxia inducible factor 1α (HIF-1α), increase survival (integrins), and reduce cell cycle arrest via FAK signaling[20]. Through these downstream ECM-activated pathways, particularly COLVI, resistance has been documented in solid tumors to multiple targeted and chemotherapeutic agents. ECM-targeted drug co-administration may provide a means to increase the efficacy of MEK inhibitors and other targeted therapies for CNs[30].
CNs APPEAR IN LATE CHILDHOOD AND CONTINUE TO DEVELOP THROUGHOUT LIFE
CNs typically begin to arise in late childhood or early adolescence and continue to accumulate throughout the lifetime, affecting 99% of adults with NF1[31,32]. Their growth is self-limited; in most patients, they reach an average maximum diameter of ~5 mm, although sizes can range from a few millimeters to 10 cm or larger. Inter-individual and interfamilial phenotypes can be striking. An 8-year prospective study revealed that a group-averaged change in CN volume among adults varies in body region but is overall quite modest - on average ~3.5 mm3 per annum, with the fastest growth seen on the back and abdomen. Similarly, the average number of new neurofibromas observed was approximately one novel lesion every 4 years during the 8-year observation period, again with the highest rate of appearance on the back and abdomen[33]. A five-year natural history study of CN accumulation is open at the National Institutes of Health, utilizing whole-body photography to track lesion development, which could provide much-needed data on predictive factors for CN initiation, growth rates, and variability (ClinicalTrials.gov ID NCT05581511).
Hormones have long been suspected to act as mitogens for cutaneous neurofibromas. This hypothesis was initially based on the observation that these tumors often appear during early adolescence, a developmental stage characterized by high circulating sex hormone levels[34]. Numerous preclinical experiments support a potential role for hormones in CN tumorigenesis and growth. CNs express hormone receptors, and Schwann-like tumor cell proliferation in vitro is buttressed by adding hormones to the growth medium[35-37]. Subjective assessments have also documented that postpartum women retrospectively perceived a faster growth rate of CNs. However, there are no compelling prospective human data suggesting that either exogenous or endogenous hormones significantly influence tumor burden or growth rate across different sexes, or among individuals receiving hormone or anti-hormone therapies. One self-report study noted that two women receiving depot contraceptives containing high levels of synthetic progesterone believed their CN growth rate increased during treatment[38]. Therefore, while there is no scientific evidence to support avoiding hormone or anti-hormone therapies altogether, women may be advised to consider alternative forms of contraception rather than depot injections or implants, particularly given similar weak associations observed with other tumors, such as meningiomas.
CN appearance and volume increase are difficult to assess both clinically and experimentally[39]. There can be a high degree of inter-investigator variability on manual measurements, confounded by the use of different analytic tools (different brands/types of calipers), different methods of measurement (tumor base vs. largest tumor girth), and the degree of pressure exerted by the calipers on these rubbery/soft, sometimes inflamed lesions. The moment of nascence of CNs is also difficult to pinpoint, partly because pools of atypical tumor-like Schwann cells in apparently normal skin could represent a microscopic/primordial form of CNs, but there is no means to determine their prospective potential for growing into a visible papule. Retrospective patient report is notably unreliable, and periodic inflammation or differences in lighting may furthermore alter the investigator’s attention to the presence or size of a tumor.
Future endeavors should strive to identify more CN measures that are independent of human error and variability. Photography, particularly 3-D photography, together with artificial intelligence (AI) has the potential to provide a universally accepted objective measure of CN total body burden, but the analytic software is designed to detect macular lesions with pigmentary differences from surrounding skin, and has not yet been optimized to automatically detect and measure voluminous and numerous idiochromatic CNs against a background of potentially abnormal surrounding skin. Furthermore, the research community would benefit from developing a standardized calibrated caliper device that applies a low, consistent pressure to CNs to eliminate inconsistencies associated with different tool sizes, manufacturers, weights, gliding friction, and manually applied pressures. It is important to learn from patients what degree of CN reduction is meaningful to them. Given that destructive treatments completely eliminate the tumor, resulting in flat skin, a drug therapy that only partially reduces the size of a CN may or may not be acceptable to the patient population as a whole.
THE GENETICS OF CN SEVERITY REMAINS POORLY UNDERSTOOD
There are many subclassifications of CNs based on their dermal vs. subdermal location, gross anatomy, and histopathology; however, the shared underlying mechanism for tumorigenesis is considered constant across different tumors and individuals: loss of the wild-type NF1 allele, leading to complete/near-complete deficiency of translated neurofibromin protein. Mutations affecting the NF1 ultrastructure or pathologic gene alterations directly targeting the Protein Kinase C (PKC) domain may impact CN severity, at least in certain ethnic populations[40]. Additional congenital polymorphisms or mutations may influence overall CN burden, whereas somatically acquired genetic alterations could contribute to individual tumor heterogeneity. There are a few causative NF1 mutations known to be associated with the phenotype and number of CNs. For example, microdeletion of the NF1 gene results in an extreme disease phenotype characterized by a heavy burden of CNs[41], while several other mutations have been identified that are associated with a reduced incidence of CNs[42-45]. However, in the vast majority of patients, a specific pathogenic germline mutation does not adequately predict CN features. The literature surrounding this question has been unequivocal that individuals within the same family with the same NF1 alteration can exhibit disparate cutaneous manifestations. In contrast to this well-accepted discordance between genotype and phenotype, the author’s clinical experience suggests that the general burden and appearance of cutaneous and subcutaneous neurofibromas within families are often similar. The generation of databases documenting quantitative features of CNs will help future research to hone in on genetic and environmental modifiers of CN clinical presentation[46].
Multiple evidence-driven hypotheses have sought to determine the cell of origin that initiates CNs through Knudson’s “Second Hit” in the remaining wild-type NF1 gene. Loss of function of the intact NF gene can occur through mutations in coding and noncoding regions as well as deletions, and are not always detectable using standard gene sequencing, which may explain why dual hits are not identified in all CNs[47,48]. Chromosomal aberrancies resulting from deletions and amplifications are commonly identified in cutaneous neurofibromas including recurrent losses in chromosomes 1, 2q, 3p, 4p, 5q, 6q, 7q, 12q, 19p, and 20p, and gains in chromosomes 2p and 8q[49].
LABORATORY MODELS OF CNs ARE IMPERFECT
Although innovations in pharmacotherapy for plexiform neurofibromas have greatly benefited from preclinical research, a truly anthromimetic animal model for CNs has only recently become available. Early genetically engineered NF1 heterozygous mouse models failed to recapitulate the characteristic symptoms of NF1[50]. Later, conditional knockout mice with biallelic NF1 deletion in specific embryonic subpopulations of neural crest-derived Schwann cells were able to reproduce certain features of NF1 but did not develop the characteristic skin tumors[51]. Spontaneous NF1-like veterinary syndromes occur in canine and bovine animals, but not in rodents[51]. It has been hypothesized that the many years required to develop tumors in humans cannot be modeled by a rapid cycling mammal with a relatively brief life expectancy, such as the mouse. Porcine models have been developed for multiple medical conditions, as pig physiology and lifespan more closely mimic that of humans. In 2018, genetically altered NF1+/R1947X Ossabaw minipigs were developed that spontaneously developed large diffuse mass-like cutaneous neurofibromas in ~40% of pigs at 4 months of age[52]. The same year, another group published a NF1+/ex42del Yucatan mini-swine model in which 44% of animals develop cutaneous tumors of the neck between 11-17 months of age[53]. Both models exhibited this non-discreet morphology with continuous enlargement in contrast to the nodular self-limited growth most common with human CN, and interestingly, the findings were mostly associated with gonadally intact male pigs, suggesting some role for androgens in CN tumorigenesis within this model.
In vitro models of CNs include primary cell culture and co-culture of Schwann-like tumor cells and
Innovative research has demonstrated that the cell of origin could reside in various sources, including multipotent bulge hair follicle cells[57], a specialized population of skin-derived precursor cells of neural crest origin[58], boundary cap cells of the dorsal root ganglia[59], dysplastic vascular pericytes[7], or deviant endoneurial or cutaneous nerve axon-associated Schwann cells - potentially through misactivation of the injury-repair pathway[60-63]. Confounding this research is the inherently plastic nature of many neural
CN tumorigenesis and ultimate dormancy are likely multifactorial, contingent upon microenvironmental contributions from haploinsufficient tumor-associated nerve, immune, and dermal cells through the release of growth factors, chemokines, and cytokines. Pleiotropic effects from germline variants in different genes may also contribute.
THERE ARE NO APPROVED PHARMACOTHERAPIES FOR CNs
Currently, there are no FDA-approved medical treatments for CNs. While surgical resection of the exophytic component combined with evacuation of the hypodermal portion and careful suturing can achieve permanent tumor control and an aesthetically acceptable appearance[64], this technique can only go so far. It does not address the ongoing development of new tumors nor the often substantial burden of existing tumors. Other destructive approaches include focused ultrasound[65], CO2 laser, radiofrequency ablation[66], photodynamic therapy[67,68], Er:YAG or Nd:YAG laser[69], and electrodessication[70]. The efficacy of these approaches varies depending on tumor size and number, tolerance for scarring/ pigmentation changes, and post-procedural pain. A number of topical and systemic medications have been tested, with the most promising results to date seen with MEK inhibitors[70-72]. Most notably, NFX-179, a topical ointment applied daily for 28 days to five medium-sized CNs (5-10 mm) in 42 subjects, resulted in a ≥ 50% reduction in tumor volume in 20% of tumors and patients. In addition, there was a 47% reduction in phosphorylated ERK levels measured via automated Western blot, with practically no adverse events. However, CN-related quality of life outcomes were not assessed in this Phase 2a trial. Overall, patient satisfaction regarding efficacy, pain, scarring, durability, and side effect profiles across all treatments evaluated to date leaves room for improvement.
A search of ClinicalTrials.gov at the time of writing identified eighteen clinical trials using the search term “cutaneous neurofibroma” in the indication field [Table 1]. Of these, the majority[14] were sponsored by academic medical centers, while the remaining four were industry-sponsored (covering three drugs). Ten trials investigated novel indications/pharmacotherapies, including three drugs targeting MEK inhibition; three different Mek inhibitors tested within 4 different trials; 2 drugs targeting mTOR, two immunotherapeutic agents, one prostaglandin inhibitor, and one vascular endothelial growth factor (VEGF) inhibitor. The remaining trials explore(d) concurrent injectable drugs with laser or photodynamic therapy. Methods to enhance drug penetration into CNs included laser microporation, microneedling, and direct intratumoral injection. At the time of writing, two trials were actively recruiting and one had not yet begun enrollment. None of these approaches had demonstrated a definitive, clinically acceptable reduction in CN size or number[73]. Several studies have evaluated patient preferences regarding treatment outcomes. Unsurprisingly, many patients indicated a willingness to take oral or topical medication over an extended period if it could reduce the number of CNs by 30% and prevent the formation of new tumors[74].
Clinical trials of cutaneous neurofibroma from
| Trial number | Study title | Status | Intervention | Mechanism |
| NCT06300502 | Assessing the efficacy of repeat, monthly treatments of cutaneous neurofibromas (cNFs) | NOT YET RECRUITING | Kybella or Asclera injection with 1064 nm Nd:YAG laser or 755 nm alexandrite laser | Destructive |
| NCT06159166 | Mirdametinib monotherapy in adults with neurofibromatosis 1 (NF1) and cNF | RECRUITING | Oral mirdametinib | MEK inhibition |
| NCT02728388 | Photodynamic therapy for benign dermal neurofibromas- phase II | RECRUITING | Topical aminolevulinic acid | Destructive |
| NCT06132165 | Efficacy of skin cooling in reducing pain associated with non-invasive treatments of neurofibromatosis type 1 cutaneous neurofibromas | RECRUITING | Deoxycholic acid or polidocanol injection with 1064 nm Nd:YAG laser or 755 nm alexandrite laser | Destructive |
| NCT05438290 | DPCP to treat cutaneous neurofibromas associated with NF1 | COMPLETED | Topical Diphencyprone | Immunotherapy |
| NCT04730583 | Tolerability of device based therapies for neurofibromatosis type 1 cutaneous neurofibromas | COMPLETED | Kybella injection with 1064 nm Nd:YAG laser or 755 nm alexandrite laser | Destructive |
| NCT05119582 | HIFU treatment of cutaneous neurofibromas in neurofibromatosis type 1: safety and efficacy | COMPLETED | TOOsonix system ONE-M device | Destructive |
| NCT05005845 | NFX-179 topical gel treatment for adults with NF1 and cNF | COMPLETED | Topical NFX-179 gel | MEK inhibition |
| NCT04435665 | NFX-179 topical gel treatment in adults with NF1 and cNF | COMPLETED | Topical NFX-179 Gel | MEK inhibition |
| NCT03090971 | Use of topical liquid diclofenac following laser microporation of cutaneous neurofibromas in patients with NF1 | COMPLETED | Laser microporation device and topical diclofenac sodium | Prostaglandin inhibition |
| NCT00865644 | Topical imiquimod 5% cream for treatment of cutaneous neurofibromas in adults with neurofibromatosis 1 | COMPLETED | Topical imiquimod 5% cream | Immunotherapy |
| NCT00657202 | Ranibizumab for neurofibromas associated with neurofibromatosis 1 | COMPLETED | Oral ranibizumab | VEGF inhibitor |
| NCT01031901 | Topical rapamycin therapy to alleviate cutaneous manifestations of tuberous sclerosis complex (TSC) and NF1 | COMPLETED | Topical sirolimus or rapamycin | mTOR inhibition |
| NCT02839720 | Selumetinib in treating patients with neurofibromatosis type 1 and cutaneous neurofibroma | COMPLETED | Oral selumetinib sulfate | MEK inhibition |
| NCT01682811 | Phase I photodynamic therapy (PDT) for benign dermal NF1 | COMPLETED | Levulan injection or topical, with or without microneedling, with photodynamic therapy | Destructive |
| NCT06120036 | Dosing and Tolerability of deoxycholic acid vs. polidocanol in the treatment of neurofibromatosis type 1 cutaneous neurofibromas | COMPLETED | Kybella injection, or Asclera injection | Destructive |
| NCT02332902 | Everolimus for treatment of disfiguring cutaneous lesions in neurofibromatosis 1 CRAD001CUS232T | COMPLETED | Oral everolimus | mTOR inhibition |
| NCT00921037 | First clinical study of erbium - yttrium aluminium garnet (YAG) laser vaporization of cutaneous neurofibromas | UNKNOWN | Erbium-YAG laser device | Destructive |
Emerging data from clinical trials of systemic MEK inhibitors suggest that this drug class may play a role in slowing or halting the development of new CNs and/or reducing the size of existing CNs[75]. Special consideration must be allotted to drug development for this benign tumor, as currently approved MEK inhibitors (mirdametinib and selumetinib) are designed to treat large, functionally impactful plexiform neurofibromas, and have a side effect profile that is unlikely to be acceptable for managing cutaneous manifestations. The primary detriment to quality of life associated with CNs derives from tumor disfigurement, while over 90% of patients on currently approved MEK inhibitors additionally experience a conspicuous acneiform rash. This visible adverse effect could exacerbate societal stigmatization and likely hinder the use of MEK inhibitors for treating CNs, despite potential benefits in tumor reduction. A 2025 study was the first to unequivocally demonstrate that systemic MEK inhibition can reduce CN tumor burden, achieving a maximum response of a 29% decrease in CN volume. However, similar to other trials, adverse events such as dry skin and rash were common[76]. It is possible that a reduced dose of MEK inhibitors, administered prophylactically or longitudinally, could mitigate these adverse events while slowing tumor growth; however, this approach has not yet been explored.
Data released in 2025 provided the most compelling evidence to date for the treatment of CNs using the topical selumetinib formulation NFX-179[72]. At the highest dose tested, CNs exhibited approximately a 50% reduction in phosphorylated ERK levels, a recognized biomarker of MEK inhibition. Decreases in tumor volume were observed in all groups, including the control group, but the highest drug dose led to an additional ~10% average reduction in tumor volume compared with the control. Most notably, around 20% of CNs treated with the highest dose of the topical formulation exhibited a ≥ 50% reduction in volume. Future generations of MEK inhibitors, as well as novel molecularly targeted therapies and topical, biologic, or injectable treatments, may further expand the arsenal of approaches to address this psychologically and socioeconomically impactful aspect of NF1. Beyond MEK inhibition, several groups are also investigating the unique transcriptional and cellular fingerprints of CNs to identify multi-target therapeutic strategies[77,78].
DISCUSSION
People with NF1 are hugely impacted by the visual appearance and physical symptoms of CNs, but current treatment options are largely limited to destructive techniques. Surgical specialties (general surgery, plastic surgery, dermatology, or neurosurgery) do not generally have the bandwidth to dedicate extensive operating room time to removing the substantial tumor burden that affects many NF1 patients. Electrodessication, although effective against small CNs measuring 5 mm or less, cannot treat larger lesions. Moreover, it is rarely offered, given the need for general anesthesia, specialized training and equipment, and its relatively low reimbursement rate compared with other procedures. Laser therapies can be very effective, but are limited by post-procedural pain. All destructive techniques carry the risk of scarring and pigmentation changes that may ultimately offset the cosmetic benefits. Neurologists and primary care doctors can help address the need for CN management by training in tumor resection, which can be performed in a clinical setting without the need for a sterile operating room. A well-vetted training course with certification by a respected institution such as the Children’s Tumor Foundation could provide the education for doctors and advanced practice providers, and a recertification course providing continuing medical education credits could ensure a durable skillset.
The human relevance of preclinical CN models has been improving over the past decade, but still requires further refinement. Porcine skin closely resembles human skin in cellular morphology, architecture, and the thickness of the four dermal layers. However, minipigs have additional cell layers in the stratum spinosum and granulosum and a higher proportion of apocrine versus eccrine sweat glands, potentially affecting topical drug diffusion. Indeed, an in vitro porcine skin permeability assay of a lipophilic compound predicted to have high skin permeability revealed striking differences: no permeability in porcine skin compared with human skin[79]. These physiologic distinctions could influence outcomes and dose determination in preclinical trials of topical medications. The skin of juvenile minipigs best replicates adult human skin[80]; however, CNs in minipig models arise in adulthood, when their skin is less biosimilar to that of humans. Thus, these models may be more appropriate for testing systemic therapies rather than topical drugs. Further basic research is needed to understand why murine and porcine models fail to spontaneously and accurately replicate CN morphology and distribution.
ECM is the major component of CNs and can block drug delivery by increasing tumor parenchymal back pressure against capillary extravasation and physically impeding large molecule diffusion. Additionally, the ECM may interfere with T cell and antibody-based therapies, as cytotoxic T cells must navigate this dense “briar patch” of ECM to reach their targets. Collagen turnover is tightly regulated through an interplay between extracellular zinc-dependent endopeptidase activity and intracellular recycling. MMPs initiate large-scale collagen breakdown by cleaving a small moiety of extracellular collagen after it binds to cell surface receptors. Internalization then occurs through phagocytosis, pinocytosis, endocytosis, or autophagocytosis, with subsequent degradation mainly within phagolysosomes by cathepsin cysteine proteases. While current CN-directed therapies aim to kill or induce senescence in tumor cells, the bulk of cutaneous neurofibromas is composed of ECM. Extracellular collagen, in the absence of enzymatic degradation by MMPs, has a longevity that dwarfs the average human life expectancy due to its dense helical structure and resistance to proteolysis. In CN tumor cells, MMP expression is downregulated in a
Future strategies should consider a dual-targeted or stepwise approach to address both the tumor cells and the persistent ECM. Potential strategies include the injection of ECM-degrading enzymes or systemic treatment with small molecules that upregulate lysosomal collagen degradation, either preceding or concurrent with Ras pathway inhibition. More attention should be given to the role of the ECM in
CONCLUSION
Developing new and better treatments for CNs is a major priority for the 0.03% of the global population affected by NF1. CNs induce anxiety and depression, negatively impact careers and relationships, cause pain and pruritus, and present a constant visual reminder of a relentlessly progressive disease despite our best current treatments. Fortunately, the NF research community has listened to patient demands, dedicating funding and focus to addressing CNs. Alongside the laborious development of pharmacotherapies and the continued refinement of animal models, access to simple surgical resections could be expanded by subsidizing and regulating training courses for tumor removal in clinical settings. The ECM represents a previously untargeted component of CNs and may also play a role in larger tumors and malignancies associated with NF1. Focusing on drug delivery at the cellular level will be key to understanding why some pharmacotherapies fail, even when bulk tumor concentrations appear sufficient to elicit a predicted response.
DECLARATIONS
Authors’ contributions
The author contributed solely to the article.
Availability of data and materials
Not applicable.
Financial support and sponsorship
None.
Conflicts of interest
Brown RM, MD, PhD, is a paid scientific advisor to Pasithea Therapeutics, which is developing a novel MEK inhibitor PAS-004.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Written informed consent for publication of the clinical images was obtained from the patients.
Copyright
© The Author(s) 2025.
REFERENCES
1. Brown R. Management of central and peripheral nervous system tumors in patients with neurofibromatosis. Curr Oncol Rep. 2023;25:1409-17.
2. Lalvani S, Brown RM. Neurofibromatosis type 1: optimizing management with a multidisciplinary approach. J Multidiscip Healthc. 2024;17:1803-17.
3. Ortonne N, Wolkenstein P, Blakeley JO, et al. Cutaneous neurofibromas: current clinical and pathologic issues. Neurology. 2018;91:S5-S13.
4. Page PZ, Page GP, Ecosse E, Korf BR, Leplege A, Wolkenstein P. Impact of neurofibromatosis 1 on quality of life: a cross-sectional study of 176 American cases. Am J Med Genet A. 2006;140:1893-8.
5. Wolkenstein P, Zeller J, Revuz J, Ecosse E, Leplège A. Quality-of-life impairment in neurofibromatosis type 1: a cross-sectional study of 128 cases. Arch Dermatol. 2001;137:1421-5.
6. Wang J, Fu J, Zhou Y, Gao D, Qing J, Yang G. Global research trends in cutaneous neurofibromas: a bibliometric analysis from 2003 to 2022. Skin Res Technol. 2024;30:e13595.
7. Friedrich RE, Holstein AF, Middendorff R, Davidoff MS. Vascular wall cells contribute to tumourigenesis in cutaneous neurofibromas of patients with neurofibromatosis type 1. A comparative histological, ultrastructural and immunohistochemical study. Anticancer Res. 2012;32:2139-58.
8. Kallionpää RA, Ahramo K, Martikkala E, et al. Mast cells in human cutaneous neurofibromas: density, subtypes, and association with clinical features in neurofibromatosis 1. Dermatology. 2022;238:329-39.
9. Kallionpää RA, Peltonen S, Le KM, et al. Characterization of immune cell populations of cutaneous neurofibromas in neurofibromatosis 1. Lab Invest. 2024;104:100285.
10. Church C, Fay CX, Kriukov E, et al. snRNA-seq of human cutaneous neurofibromas before and after selumetinib treatment implicates role of altered Schwann cell states, inter-cellular signaling, and extracellular matrix in treatment response. Acta Neuropathol Commun. 2024;12:102.
11. McLean DT, Meudt JJ, Lopez Rivera LD, et al. Single-cell RNA sequencing of neurofibromas reveals a tumor microenvironment favorable for neural regeneration and immune suppression in a neurofibromatosis type 1 porcine model. Front Oncol. 2023;13:1253659.
12. Nagrani NS, Bhawan J. Histopathological variants of cutaneous neurofibroma: a compendious review. Dermatopathology. 2022;10:1-19.
13. Peltonen J, Penttinen R, Larjava H, Aho HJ. Collagens in neurofibromas and neurofibroma cell cultures. Ann N Y Acad Sci. 1986;486:260-70.
14. Brosseau JP, Sathe AA, Wang Y, et al. Human cutaneous neurofibroma matrisome revealed by single-cell RNA sequencing. Acta Neuropathol Commun. 2021;9:11.
15. Jiang C, McKay RM, Le LQ. Tumorigenesis in neurofibromatosis type 1: role of the microenvironment. Oncogene. 2021;40:5781-7.
16. Harigai R, Sakai S, Nobusue H, et al. Tranilast inhibits the expression of genes related to epithelial-mesenchymal transition and angiogenesis in neurofibromin-deficient cells. Sci Rep. 2018;8:6069.
17. Chen P, Cescon M, Megighian A, Bonaldo P. Collagen VI regulates peripheral nerve myelination and function. FASEB J. 2014;28:1145-56.
18. Chen P, Cescon M, Zuccolotto G, et al. Collagen VI regulates peripheral nerve regeneration by modulating macrophage recruitment and polarization. Acta Neuropathol. 2015;129:97-113.
19. Cescon M, Gregorio I, Eiber N, et al. Collagen VI is required for the structural and functional integrity of the neuromuscular junction. Acta Neuropathol. 2018;136:483-99.
21. Cescon M, Rampazzo E, Bresolin S, et al. Collagen VI sustains cell stemness and chemotherapy resistance in glioblastoma. Cell Mol Life Sci. 2023;80:233.
22. Jiang C, Kumar A, Yu Z, et al. Basement membrane proteins in extracellular matrix characterize NF1 neurofibroma development and response to MEK inhibitor. J Clin Invest. 2023:133.
23. Williams L, Layton T, Yang N, Feldmann M, Nanchahal J. Collagen VI as a driver and disease biomarker in human fibrosis. FEBS J. 2022;289:3603-29.
24. Wang WN, Koguchi-Yoshioka H, Nimura K, et al. Distinct transcriptional profiles in the different phenotypes of neurofibroma from the same subject with neurofibromatosis 1. J Invest Dermatol. 2024;144:133-141.e4.
25. Castagnaro S, Gambarotto L, Cescon M, Bonaldo P. Autophagy in the mesh of collagen VI. Matrix Biol. 2021;100-101:162-72.
26. Silver FH. The role of connections between cellular and tissue mechanical elements and the importance of applied energy in mechanotransduction in cancerous tissue. Biomolecules. 2025;15:457.
27. Lu P, Chen Z, Wu M, et al. Type I collagen extracellular matrix facilitates nerve regeneration via the construction of a favourable microenvironment. Burns Trauma. 2024;12:tkae049.
28. Li X, Shepard HM, Cowell JA, et al. Parallel accumulation of tumor hyaluronan, collagen, and other drivers of tumor progression. Clin Cancer Res. 2018;24:4798-807.
29. Rømer AMA, Thorseth ML, Madsen DH. Immune modulatory properties of collagen in cancer. Front Immunol. 2021;12:791453.
30. Henke E, Nandigama R, Ergün S. Extracellular matrix in the tumor microenvironment and its impact on cancer therapy. Front Mol Biosci. 2019;6:160.
31. Huson SM, Harper PS, Compston DA. Von Recklinghausen neurofibromatosis: a clinical and population study in south-east Wales. Brain. 1988;111:1355-81.
32. Duong TA, Bastuji-Garin S, Valeyrie-Allanore L, Sbidian E, Ferkal S, Wolkenstein P. Evolving pattern with age of cutaneous signs in neurofibromatosis type 1: a cross-sectional study of 728 patients. Dermatology. 2011;222:269-73.
33. Cannon A, Chen MJ, Li P, et al. Cutaneous neurofibromas in neurofibromatosis type I: a quantitative natural history study. Orphanet J Rare Dis. 2018;13:31.
35. Geller M, Mezitis SG, Nunes FP, et al. Progesterone and estrogen receptors in neurofibromas of patients with NF1. Clin Med Pathol. 2008;1:93-7.
36. Fishbein L, Zhang X, Fisher LB, et al. In vitro studies of steroid hormones in neurofibromatosis 1 tumors and Schwann cells. Mol Carcinog. 2007;46:512-23.
37. Pennanen P, Peltonen S, Kallionpää RA, Peltonen J. The effect of estradiol, testosterone, and human chorionic gonadotropin on the proliferation of Schwann cells with NF1+/- or NF1-/- genotype derived from human cutaneous neurofibromas. Mol Cell Biochem. 2018;444:27-33.
38. Lammert M, Mautner VF, Kluwe L. Do hormonal contraceptives stimulate growth of neurofibromas? BMC Cancer. 2005;5:16.
39. Cannon A, Jarnagin K, Korf B, et al. Clinical trial design for cutaneous neurofibromas. Neurology. 2018;91:S31-7.
40. Zhu B, Zheng T, Wang W, et al. Genotype-phenotype correlations of neurofibromatosis type 1: a cross-sectional study from a large Chinese cohort. J Neurol. 2024;271:1893-900.
41. Kehrer-Sawatzki H, Kluwe L, Salamon J, et al. Clinical characterization of children and adolescents with NF1 microdeletions. Childs Nerv Syst. 2020;36:2297-310.
42. Pinna V, Lanari V, Daniele P, et al. p.Arg1809Cys substitution in neurofibromin is associated with a distinctive NF1 phenotype without neurofibromas. Eur J Hum Genet. 2015;23:1068-71.
43. Jiang C, McKay RM, Lee SY, et al. Cutaneous neurofibroma heterogeneity: factors that influence tumor burden in neurofibromatosis type 1. J Invest Dermatol. 2023;143:1369-77.
44. Upadhyaya M, Huson SM, Davies M, et al. An absence of cutaneous neurofibromas associated with a 3-bp inframe deletion in exon 17 of the NF1 gene (c.2970-2972 delAAT): evidence of a clinically significant NF1 genotype-phenotype correlation. Am J Hum Genet. 2007;80:140-51.
45. Trevisson E, Morbidoni V, Forzan M, et al. The Arg1038Gly missense variant in the NF1 gene causes a mild phenotype without neurofibromas. Mol Genet Genomic Med. 2019;7:e616.
46. Lin MJ, Yao H, Vera K, et al. Cutaneous neurofibromas and quality of life in adults with neurofibromatosis type 1. JAMA Dermatol. 2024;160:1091-8.
47. Thomas L, Spurlock G, Eudall C, et al. Exploring the somatic NF1 mutational spectrum associated with NF1 cutaneous neurofibromas. Eur J Hum Genet. 2012;20:411-9.
48. Emmerich D, Zemojtel T, Hecht J, et al. Somatic neurofibromatosis type 1 (NF1) inactivation events in cutaneous neurofibromas of a single NF1 patient. Eur J Hum Genet. 2015;23:870-3.
49. Asai A, Karnan S, Ota A, et al. High-resolution 400K oligonucleotide array comparative genomic hybridization analysis of neurofibromatosis type 1-associated cutaneous neurofibromas. Gene. 2015;558:220-6.
50. Jacks T, Shih TS, Schmitt EM, Bronson RT, Bernards A, Weinberg RA. Tumour predisposition in mice heterozygous for a targeted mutation in Nf1. Nat Genet. 1994;7:353-61.
51. Osum SH, Watson AL, Largaespada DA. Spontaneous and engineered large animal models of neurofibromatosis type 1. Int J Mol Sci. 2021;22:1954.
52. Isakson SH, Rizzardi AE, Coutts AW, et al. Genetically engineered minipigs model the major clinical features of human neurofibromatosis type 1. Commun Biol. 2018;1:158.
53. White KA, Swier VJ, Cain JT, et al. A porcine model of neurofibromatosis type 1 that mimics the human disease. JCI Insight. 2018;3:120402.
54. Mo J, Anastasaki C, Chen Z, et al. Humanized neurofibroma model from induced pluripotent stem cells delineates tumor pathogenesis and developmental origins. J Clin Invest. 2021;131:139807.
55. Mazuelas H, Magallón-Lorenz M, Fernández-Rodríguez J, et al. Modeling iPSC-derived human neurofibroma-like tumors in mice uncovers the heterogeneity of Schwann cells within plexiform neurofibromas. Cell Rep. 2022;38:110385.
56. Liao CP, Pradhan S, Chen Z, Patel AJ, Booker RC, Le LQ. The role of nerve microenvironment for neurofibroma development. Oncotarget. 2016;7:61500-8.
57. Wu J, Williams JP, Rizvi TA, et al. Plexiform and dermal neurofibromas and pigmentation are caused by Nf1 loss in desert hedgehog-expressing cells. Cancer Cell. 2008;13:105-16.
58. Le LQ, Shipman T, Burns DK, Parada LF. Cell of origin and microenvironment contribution for NF1-associated dermal neurofibromas. Cell Stem Cell. 2009;4:453-63.
59. Radomska KJ, Coulpier F, Gresset A, et al. Cellular origin, tumor progression, and pathogenic mechanisms of cutaneous neurofibromas revealed by mice with Nf1 knockout in boundary cap cells. Cancer Discov. 2019;9:130-47.
60. Brosseau JP, Pichard DC, Legius EH, et al. The biology of cutaneous neurofibromas: consensus recommendations for setting research priorities. Neurology. 2018;91:S14-20.
61. Ge LL, Xing MY, Zhang HB, Wang ZC. Neurofibroma development in neurofibromatosis type 1: insights from cellular origin and schwann cell lineage development. Cancers. 2022;14:4513.
62. Zheng H, Chang L, Patel N, et al. Induction of abnormal proliferation by nonmyelinating schwann cells triggers neurofibroma formation. Cancer Cell. 2008;13:117-28.
63. Parrinello S, Noon LA, Harrisingh MC, et al. NF1 loss disrupts Schwann cell-axonal interactions: a novel role for semaphorin 4F. Genes Dev. 2008;22:3335-48.
64. Chamseddin BH, Hernandez L, Solorzano D, Vega J, Le LQ. Robust surgical approach for cutaneous neurofibroma in neurofibromatosis type 1. JCI Insight. 2019;5:128881.
65. Wozniak B, Bove T, Zawada T, Calik J. Treatment of cutaneous neurofibromas in patients with neurofibromatosis type 1. Case Rep Dermatol. 2023;15:194-201.
66. Yoshida Y, Sato N, Furumura M, Nakayama J. Treatment of pigmented lesions of neurofibromatosis 1 with intense pulsed-radio frequency in combination with topical application of vitamin D3 ointment. J Dermatol. 2007;34:227-30.
67. Quirk B, Olasz E, Kumar S, Basel D, Whelan H. Photodynamic therapy for benign cutaneous neurofibromas using aminolevulinic acid topical application and 633 nm red light illumination. Photobiomodul Photomed Laser Surg. 2021;39:411-7.
68. Peltonen S, Jannic A, Wolkenstein P. Treatment of cutaneous neurofibromas with carbon dioxide laser: technique and patient experience. Eur J Med Genet. 2022;65:104386.
69. Chamseddin BH, Le LQ. Management of cutaneous neurofibroma: current therapy and future directions. Neurooncol Adv. 2020;2:i107-16.
70. Levine SM, Levine E, Taub PJ, Weinberg H. Electrosurgical excision technique for the treatment of multiple cutaneous lesions in neurofibromatosis type I. J Plast Reconstr Aesthet Surg. 2008;61:958-62.
71. Verma SK, Riccardi VM, Plotkin SR, et al. Considerations for development of therapies for cutaneous neurofibroma. Neurology. 2018;91:S21-30.
72. Sarin KY, Bradshaw M, O’Mara C, et al. Effect of NFX-179 MEK inhibitor on cutaneous neurofibromas in persons with neurofibromatosis type 1. Sci Adv. 2024;10:eadk4946.
73. Ly I, Romo CG, Gottesman S, et al. Target product profile for cutaneous neurofibromas: clinical trials to prevent, arrest, or regress cutaneous neurofibromas. J Invest Dermatol. 2023;143:1388-96.
74. Guiraud M, Bouroubi A, Beauchamp R, et al. Cutaneous neurofibromas: patients’ medical burden, current management and therapeutic expectations: results from an online European patient community survey. Orphanet J Rare Dis. 2019;14:286.
75. Passos J, Soares MP, Salgado D, et al. A single-center case study series assessing the effect of selumetinib use in patients with neurofibromatosis-related plexiform neurofibromas. Neurooncol Adv. 2024;6:vdae177.
76. Gross AM, Reid OH, Baldwin LA, et al. Treatment of cutaneous neurofibromas in neurofibromatosis type 1 with mek inhibitor selumetinib: a nonrandomized clinical trial. JAMA Dermatol. 2025;161:533-7.
77. Banerjee J, Allaway RJ, Taroni JN, et al. Integrative analysis identifies candidate tumor microenvironment and intracellular signaling pathways that define tumor heterogeneity in NF1. Genes. 2020;11:226.
78. Mazuelas H, Magallón-Lorenz M, Uriarte-Arrazola I, et al. Unbalancing cAMP and Ras/MAPK pathways as a therapeutic strategy for cutaneous neurofibromas. JCI Insight. 2024:9.
79. In M, Richardson KC, Loewa A, Hedtrich S, Kaessmeyer S, Plendl J. Histological and functional comparisons of four anatomical regions of porcine skin with human abdominal skin. Anat Histol Embryol. 2019;48:207-17.
80. Liu Y, Chen JY, Shang HT, et al. Light microscopic, electron microscopic, and immunohistochemical comparison of Bama minipig (Sus scrofa domestica) and human skin. Comp Med. 2010;60:142-8.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Issue
Copyright

Data & Comments
Data
0











Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].