

Synergistic anionic and cationic co-doping in cobalt hydroxide for enhanced hydrogen production coupled with urea electrooxidation
 0
0 Abstract
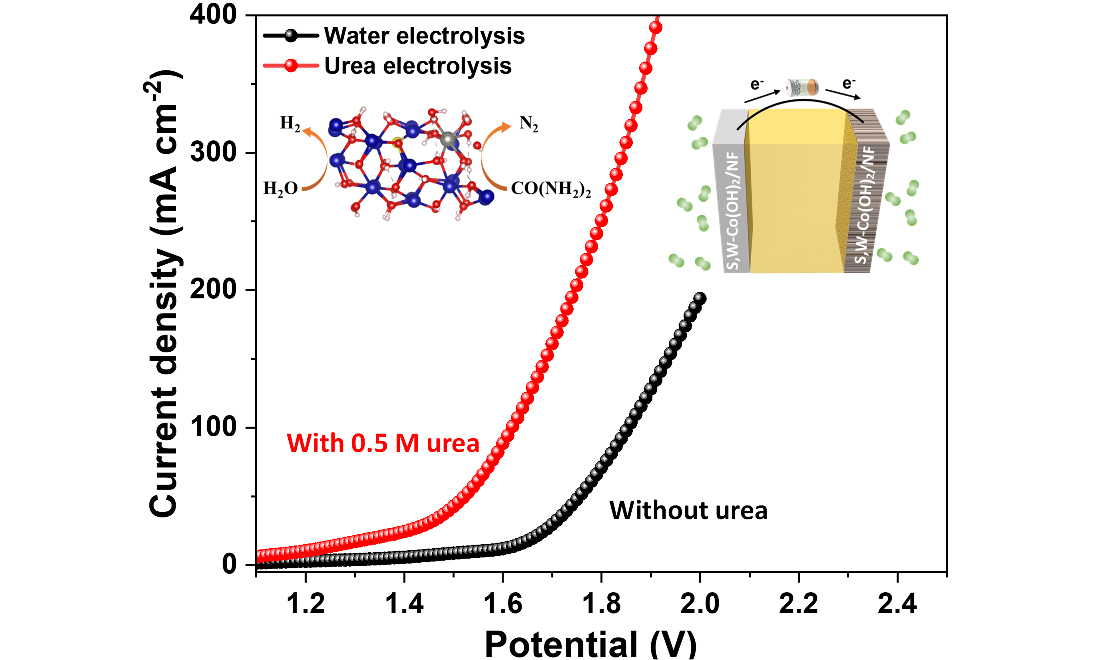
Urea-assisted electrolysis offers an energy-efficient and sustainable approach to industrial-scale hydrogen generation by reducing cell voltage relative to conventional water electrolysis. However, the design of bifunctional catalysts that deliver high activity and durability for both half-reactions remains a significant challenge. Here, we adjust the electronic structure of cobalt hydroxide via synergistic co-doping with tungsten cations and sulfur anions, optimizing the reaction pathways for hydrogen evolution reaction (HER) and urea oxidation reaction (UOR). Sulfur and tungsten co-doped cobalt hydroxide nanosheet arrays grown directly on Ni foam [S,W-Co(OH)2/NF] were prepared through a scalable one-step electrodeposition method and demonstrate outstanding catalytic performance, delivering HER overpotentials of 70 and 136 mV at current densities of 10 and 100 mA cm-2, respectively, while achieving a UOR current density of 500 mA cm-2 at 1.45 V. In a two-electrode system, the total energy consumption of urea-aided electrolysis is reduced by approximately 23% relative to conventional water electrolysis. Density functional theory (DFT) calculations reveal that dual doping enhances the electronic environment of cobalt active sites and promotes advantageous adsorption of reaction intermediates. This work presents a cost-effective strategy that couples energy-efficient hydrogen production with urea-rich wastewater treatment.
Keywords
INTRODUCTION
Hydrogen is widely recognized as a clean energy carrier owing to its high gravimetric energy density and the absence of carbon emissions[1,2]. In comparison to fossil-fuel reforming, electrocatalytic water splitting provides a sustainable method for green hydrogen production when powered by renewable energy[3]. The intrinsically slow kinetics of the hydrogen evolution reaction (HER) and the oxygen evolution reaction (OER) at the electrodes significantly limit overall energy conversion efficiency[4,5]. Substituting the OER with an alternative anodic reaction featuring a lower thermodynamic potential is an effective approach to reduce cell voltage and improve energy efficiency[6]. The urea oxidation reaction (UOR) is particularly attractive due to its low thermodynamic potential (0.37 V) and the widespread availability of urea-rich wastewater as a potentially cost-free feedstock[7-9]. However, the UOR proceeds through a complicated six-electron transfer pathway with several adsorbed intermediates, leading to sluggish kinetics despite favorable thermodynamics[10]. In alkaline media, the UOR typically proceeds via stepwise dehydrogenation of urea, generating surface-bound intermediates such as *NH2, *NH, and *N species. Subsequent C-N bond cleavage and coupling processes lead to the formation of *CO-containing species and key intermediates such as *NCO and *NCOO. The formation and transformation of these adsorbed intermediates are generally considered kinetically demanding steps, contributing to the sluggish reaction kinetics of the UOR[11, 12]. Therefore, the rational design and synthesis of electrocatalysts are urgently required to enhance the bifunctional performance toward both the HER and UOR, despite significant ongoing challenges[9,13].
Precious-metal catalysts (Pt, RuO2) exhibit optimal kinetics for water electrolysis but are hindered by scarcity, cost, and reduced durability under harsh alkaline conditions[4,14]. This has driven the transition toward earth-abundant transition metal-based materials and the coupling of electrolysis with small-molecule oxidation (e.g., urea) to reduce cell voltages[15,16]. Among transition metal compounds, transition metal hydroxides (TMHs) are notable for their low cost, ease of synthesis, and robust alkaline stability, yet their inadequate conductivity and restricted intrinsic activity limit practical applications. From the perspective of electronic structure modulation, the key to alkaline HER is tuning the d-band center of active metal sites to balance H2O dissociation and H* adsorption. This can be achieved through heteroatom doping or defect engineering, as demonstrated in Ni- and Co-based hydroxides, which exhibit improved activity and durability[17-20]. Conversely, the UOR involves sluggish multistep dehydrogenation, competing OER, and intermediate poisoning, necessitating precise control over surface charge distribution and intermediate binding[21,22]. Targeted anion or cation doping has emerged as an effective route to modulate charge distribution and intermediate binding, thereby improving UOR onset and durability on Co/Ni hydroxides and spinel-like structures[23-25]. For instance, V-doped Co2P4O12 and NiFeCo-LDH/NF exhibit improved bifunctional performance via dopant-induced charge redistribution and protective surface reconstruction[26,27]. Despite these advances, designing a single catalyst that efficiently drives both HER and UOR remains challenging due to conflicting intermediate adsorption requirements and stability issues at high current densities. These considerations motivate our strategy to develop a dual-doped cobalt hydroxide catalyst to synergistically tune the Co-centered electronic structure and achieve robust bifunctional activity for overall water and urea electrolysis.
Based on the aforementioned strategy for optimizing the electronic configuration of active sites through doping engineering, we designed and synthesized sulfur (S) and tungsten (W) co-doped cobalt hydroxide supported on nickel foam [S,W-Co(OH)2/NF]. The dual dopants synergistically modulate the electronic configuration of Co(OH)2 and enhance its electrical conductivity, while the hierarchical nanosheet architecture facilitates rapid mass transport and efficient charge transfer. In alkaline media, the dual-doped catalyst exhibits minimal overpotentials at practical current densities for the HER. In a urea-containing electrolyte, it efficiently drives the UOR at relatively low potentials, sustaining stable operation at high currents over prolonged periods. In a two-electrode design, the urea-supported electrolyzer achieves high current densities at significantly lower cell voltages than conventional water electrolysis under identical conditions. DFT calculations indicate that dopant-metal orbital interactions influence the electronic structure, leading to a near-thermoneutral hydrogen adsorption free energy (ΔGH∗) for hydrogen adsorption and reduced energy barriers for urea dehydrogenation. These findings collectively identify S,W-Co(OH)2/NF as an efficient and robust bifunctional catalyst for HER and UOR, presenting a viable approach toward energy-efficient hydrogen production coupled with urea oxidation.
EXPERIMENTAL
Chemicals
All reagents employed in this study were used as received without any additional purification. Cobalt nitrate hexahydrate (Co(NO3)2·6H2O, reagent grade 99%), ammonium fluoride (NH4F, reagent grade 96%), and hydrochloric acid (HCl) were acquired from Aladdin Industrial Corporation (Shanghai, China). Thiourea (CH4N2S, reagent grade 99%), tungsten (VI) chloride (WCl6, reagent grade 99%), a 5 wt.% Nafion solution, and potassium hydroxide (KOH, reagent grade 90%) were obtained from Macklin (Shanghai, China). A commercial Pt/C catalyst (20 wt.%) was supplied by Hessen Corporation (Shanghai, China).
Catalyst preparation
A one-step electrodeposition strategy was used to prepare S,W-Co(OH)2/NF. In a standard fabrication, a fragment of NF substrate (1 × 1 cm2) was thoroughly washed in 3.0 M HCl, distilled water, and anhydrous ethanol multiple times to remove surface oxides and organic contaminants. Electrodeposition was conducted at room temperature in a two-electrode system using 50 mL of electrolyte containing 4 mM Co(NO3)2·6H2O, 20 mM CH4N2S, 13.5 mM NH4F, and 0.05 mM WCl6. The pretreated NF served as the working electrode, while a graphite rod served as both the counter electrode and reference electrode. Deposition was performed at -5 V for 30 min, followed by rinsing with water to obtain the S,W0.05-Co(OH)2/NF electrode. Dual-doped catalysts with W0.01 and W0.1 were synthesized using the same method, with WCl6 concentrations adjusted to 0.01 mM and 0.1 mM, respectively. Samples with different deposition times (10, 20, and 30 min) were also prepared for comparison. The S-Co(OH)2/NF sample was fabricated in the same way as the dual-doped catalyst, but without the addition of WCl6 to the electrolyte. Similarly, W-Co(OH)2/NF was synthesized by omitting CH4N2S from the electrolyte. For the Pt/C/NF electrode, commercial 20% Pt/C powder was dispersed in a mixed solution of deionized water, ethanol, and Nafion binder, and then drop-cast uniformly onto the NF surface. The resulting Pt/C/NF was then dried overnight in a vacuum oven.
Catalyst characterization
The morphology and structural features of the synthesized products were characterized by field emission scanning electron microscopy (FESEM; SU-8220, Hitachi, Tokyo, Japan) and Transmission electron microscopy (TEM; HT7700, JEM-2010, Talos F200X, Hitachi, Tokyo, Japan). Powder X-ray diffraction (XRD; TTR-III, Tokyo, Japan) analysis was performed to determine the crystalline structure. Raman spectra were recorded using a Renishaw inVia Raman spectrometer with a 532 nm laser as the excitation source. Energy-dispersive spectroscopy (EDS) mapping was conducted using a Talos F200X (Thermo Fisher Scientific, USA) TEM working at 200 kV. Surface chemical states were analyzed by X-ray photoelectron spectroscopy (XPS; ESCALAB 250, UK) using Al Kα radiation as the excitation source.
Electrochemical measurements
The electrochemical performance of the synthesized catalysts was evaluated using a three-electrode system with a Corrtest (CS-310X) electrochemical workstation at ambient temperature. The prepared samples functioned as the working electrode (testing area: 0.25 cm2), whereas Hg/HgO (1 M KOH) served as the reference electrode, and a graphite rod was used as the counter electrode in an alkaline medium. The HER and UOR measurements were conducted in 1.0 M KOH and 1.0 M KOH containing urea, respectively. The linear sweep voltammetry (LSV) curves were obtained at 5 mV s-1, and the Tafel slope was derived by fitting the linear region of the Tafel plot using the Tafel equation, [η = b log (j) + a]. Electrochemical impedance spectroscopy (EIS) was performed over a frequency range from 100 kHz to 0.01 Hz with an amplitude of 5 mV. The electrochemically active surface area (ECSA) was calculated according to a previously reported equation: ECSA = Ageo × Cdl /Cs, where Ageo is the geometric area of the electrode, Cdl represents the electric double-layer capacitance in the non-Faradaic region, and Cs is the specific capacitance of the material (typically 0.04 mF cm-2). Cdl was determined from cyclic voltammetry (CV) measurements at scan rates ranging from 10 to 50 mV s-1. The current density difference (Δj = ja - jc) was graphed against the scan rate, and the slope of the linear fit corresponds to twice (Cdl). All potentials measured during the three-electrode performance tests were iR-corrected and converted to the reversible hydrogen electrode (RHE) scale using the following equation: E(RHE) = E(Hg/HgO) + 0.059 × pH + 0.098 - 90% × iR
Computational methods
This study utilized the Vienna Ab Initio Simulation Package (VASP) for theoretical computations of materials, including DFT[28]. The Perdew-Burke-Ernzerhof (PBE) exchange-correlation functional was adopted within the generalized gradient approximation (GGA), together with projector augmented-wave (PAW) pseudopotentials[29]. The empirical dispersion-corrected DFT method, specifically DFT-D3, was employed to accurately account for weak long-range van der Waals interactions. A kinetic energy cutoff of 500 eV was applied for the plane wave expansion. The convergence criterion for the self-consistent field (SCF) iterations was set to 10-5 eV. Brillouin zone integration was executed using the Gamma sampling technique, with a geometric optimization density of 2 × 2 × 1 and an electronic structure calculation density of 4 × 4 × 1. A vacuum region of 15 Å was implemented in all systems to prevent interactions between adjacent slabs. The geometric optimization was performed using the conjugate gradient method, ensuring that the force on each atom remained below 0.03 eV Å-1. The Gibbs free energy change for adsorption or reaction processes was calculated as: ΔG = ΔE + (ZPE-TΔS), where ΔE is the adsorption or reaction energy derived from DFT calculations, ΔZPE indicates the zero-point energy correction, T represents the temperature, and ΔS is the entropy change.
RESULTS AND DISCUSSION
Preparation and structural analysis
[Figure 1A] schematically illustrates the facile one-step electrodeposition used to fabricate
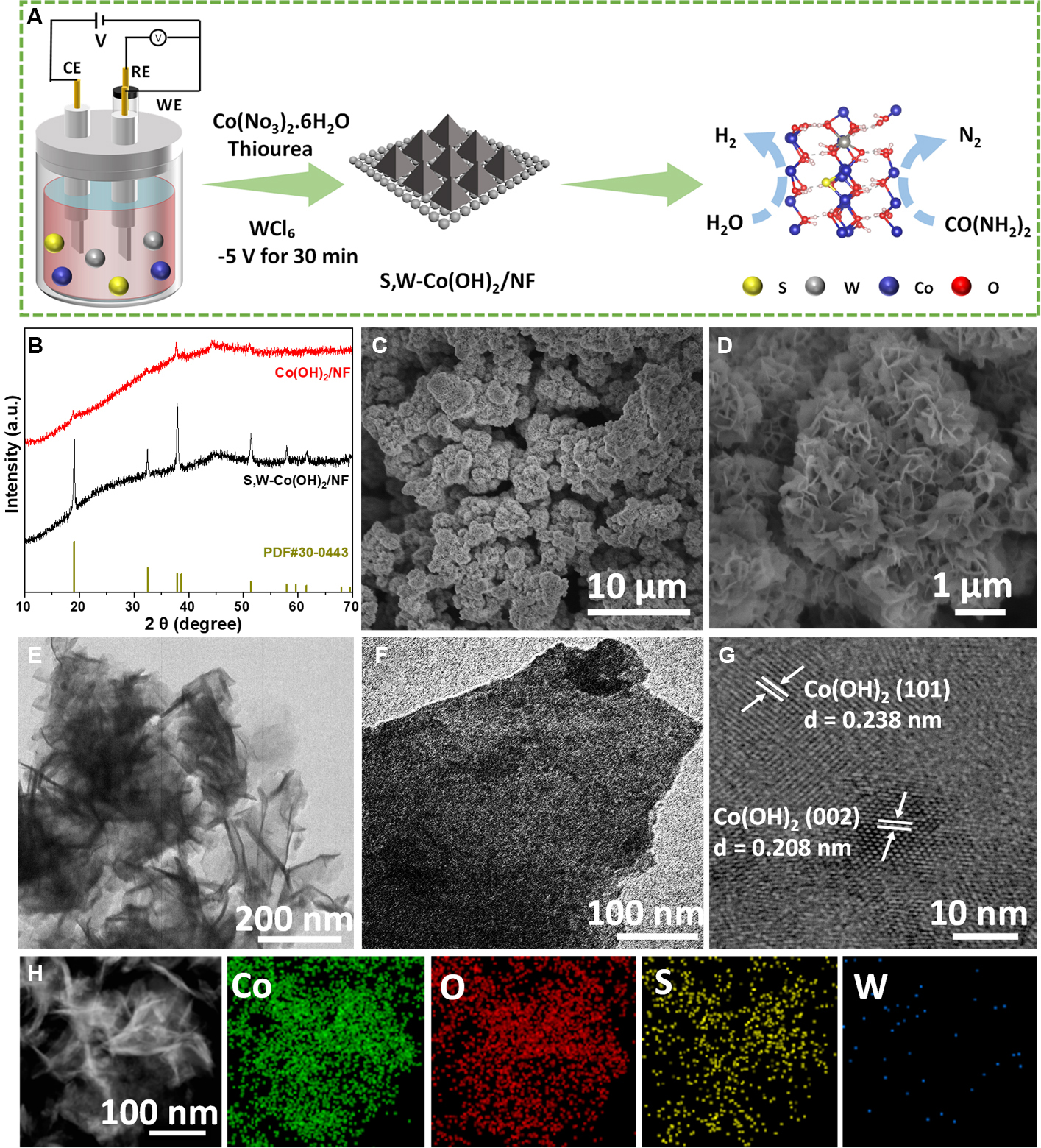
Figure 1. (A) Schematic illustration of S,W-Co(OH)2/NF; (B) XRD analysis of Co(OH)2/NF and S,W-Co(OH)2/NF; (C and D) SEM images; (E and F) TEM images; (G) HRTEM analysis of S,W-Co(OH)2/NF; (H) EDS with corresponding elemental mapping images. CE: Counter electrode; RE: reference electrode; WE: working electrode; NF: nickel foam; XRD: X-ray diffraction; SEM: scanning electron microscopy; TEM: transmission electron microscopy; HRTEM: high-resolution transmission electron microscopy; EDS: energy-dispersive spectroscopy.
S,W-Co(OH)2/NF. The single-doped and pristine catalysts were synthesized under identical conditions but without the dopant sources. After deposition, the initially porous NF, as depicted in [Supplementary Figure 1], is uniformly covered by precursor nanosheets, as shown in [Supplementary Figure 2], forming a conformal and porous catalyst layer. The XRD patterns in [Figure 1B] confirm that both catalysts adopt the Co(OH)2 phase (JCPDS No. 30-0443). Although slight peak broadening is observed after doping, the characteristic reflections of Co(OH)2 are retained, indicating a preserved phase structure. The lattice distortion induced by W and S incorporation enhances defect density and electronic conductivity, contributing to improved electrocatalytic performance. Likewise, S-Co(OH)2/NF and W-Co(OH)2/NF display characteristic diffraction patterns of Co(OH)2, as depicted in [Supplementary Figure 3]. Diffraction peaks from metallic nickel arise from the Ni foam substrate. The morphology of the samples was investigated by FESEM and TEM. As presented in [Figure 1C and D], the FESEM images show a three-dimensional network of interconnected nanosheets, which provides numerous exposed active sites. TEM further corroborates the ultrathin morphology of dual-doped S,W-Co(OH)2/NF, as demonstrated in [Figure 1E and F]. The HRTEM image of S,W-Co(OH)2/NF in [Figure 1G] reveals distinct lattice fringes with d-spacings of 0.238 and 0.208 nm, corresponding to the (101) and (002) planes of Co(OH)2, respectively, consistent with the XRD results. Furthermore, the EDS mapping data depicted in [Figure 1H] demonstrate homogeneous elemental distributions of Co, oxygen (O), S, and W across the nanosheets, underscoring the efficacy of the doping process. Although the W signal in EDS appears relatively weak due to its low concentration, inductively coupled plasma (ICP) analysis confirms a W content of 0.73 at% [Supplementary Figure 4 and Supplementary Table 1], verifying successful incorporation. XPS further confirms the chemical state of W within the lattice.
Analysis of composition and atomic structure
Raman spectroscopy, owing to its sensitivity to subtle microstructural variations in inorganic frameworks, was employed to probe the lattice characteristics of both catalysts. As shown in [Figure 2A], both samples exhibit nearly identical peaks, indicating that W incorporation preserves the overall Co(OH)2 lattice. Nevertheless, S,W-Co(OH)2/NF displays discernible red shifts in the vibrational bands centered at 466, 505, and 660 cm-1 relative to S-Co(OH)2/NF, indicative of lattice perturbation and modified local bonding environments upon W incorporation, consistent with vibrational softening around Co sites[30]. XPS was performed to clarify the surface composition and valence states of the pristine, S-doped, and dual-doped catalysts. The survey spectrum of the dual-doped catalyst [Figure 2B] validates the existence of Co, S, O, and W. The Co 2p spectrum was deconvoluted into two spin-orbit doublets with associated satellite peaks, yielding Co3+ peaks at 781.0 and 796.9 eV and Co2+ peaks at 783.0 and 797.8 eV, as shown in [Figure 2C][31]. Notably, the Co 2p binding energies of the dual-doped catalyst shift positively by 0.34 eV in comparison to the S-doped and pristine catalysts, evidencing electron redistribution and a modified Co electronic structure induced by W doping. The high-resolution S 2p spectra [Figure 2D] for both catalysts exhibit three characteristic peaks corresponding to S 2p3/2, S 2p1/2, and S-O[32]. The W 4f spectrum [Figure 2E] exhibits two distinct peaks at binding energies of 35.1 eV and 37.2 eV, attributed to the W 4f7/2 and W 4f5/2 spin-orbit components, respectively, corroborating the existence of W6+[33]. Although the surface W concentrations appear comparable, the catalytic performance is governed by electronic structure modulation rather than dopant content alone. The synergistic interaction between W and S induces more effective charge redistribution and optimized adsorption energetics, leading to enhanced catalytic activity. In the O 1s XPS spectra [Figure 2F], the catalysts exhibit three components located at 530.5, 531.5, and 533.0 eV, corresponding to lattice oxygen (M-O), hydroxyl species (M-OH), and adsorbed H2O, respectively. The dominant M-OH peak indicates a hydroxyl-rich surface, while slight intensity variations after S and W doping suggest modulation of the surface oxygen environment[34,35]. The C 1s spectrum [Supplementary Figure 5] displays three characteristic carbon peaks at 284.8, 285.4, and 288.4 eV, corresponding to C-C, C-O-C, and C=O species, respectively. All three samples exhibit similar features, which are attributed to adventitious carbon and surface residues arising from air exposure or synthesis-related residues[36]. Collectively, the XPS findings confirm the effective incorporation of S and W into Co(OH)2/NF and reveal associated electronic structure modulation and local coordination changes, which are beneficial for enhancing bifunctional HER and UOR electrocatalytic performance.
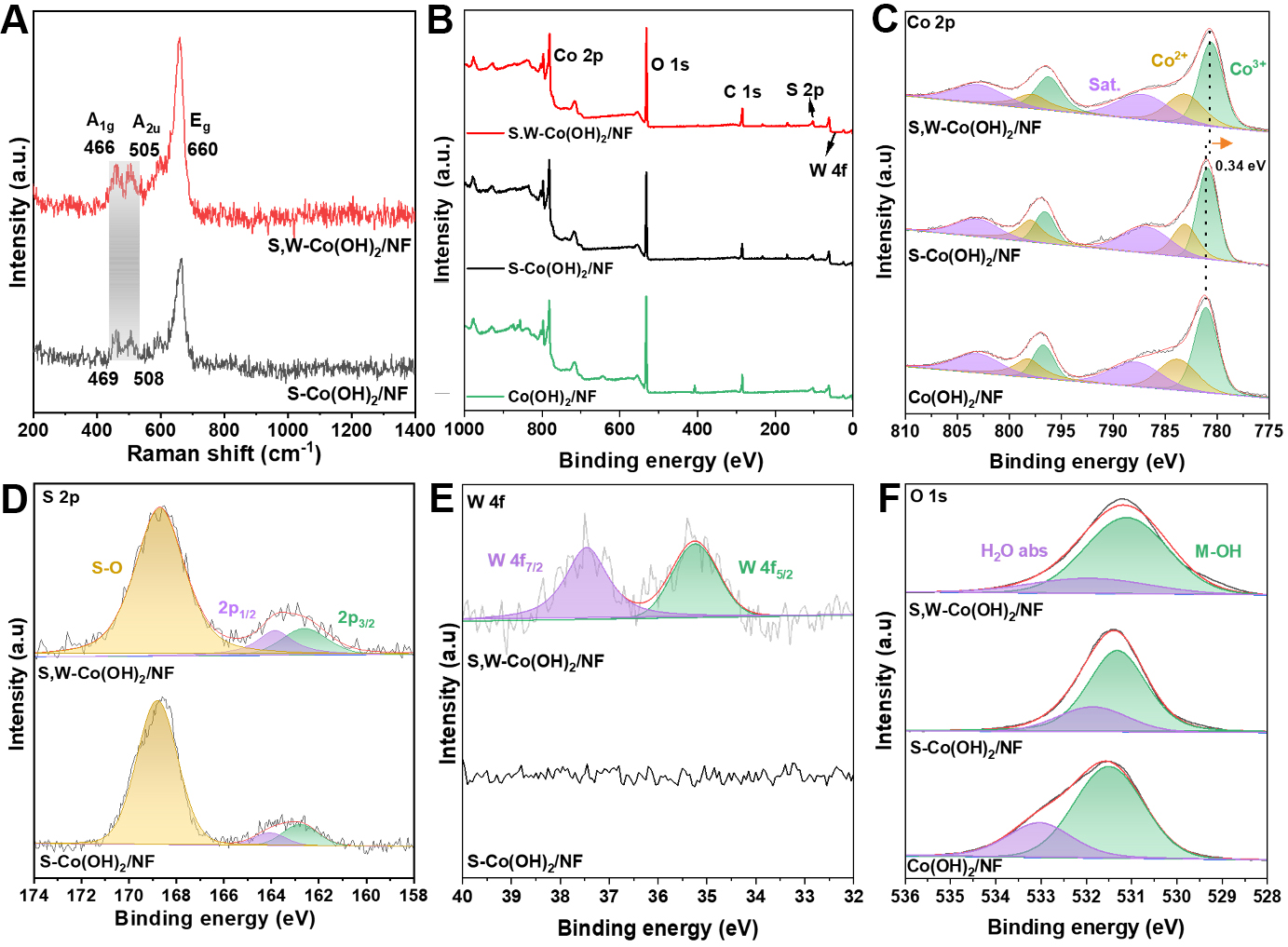
Figure 2. (A) Raman spectra of the synthesized catalysts; (B) XPS survey spectrum and high-resolution XPS spectra of (C) Co 2p; (D) S 2p; (E) W 4f; and (F) O 1s. NF: Nickel foam; XPS: X-ray photoelectron spectroscopy.
Electrocatalytic HER
The HER activity was initially evaluated in 1.0 M KOH using a three-electrode setup at room temperature. LSV reveals clear performance differences among the fabricated samples, as illustrated in [Figure 3A]. The LSV results suggest that bare NF demonstrates poor HER performance, while the single-doped and pristine samples display reasonable performance levels. In contrast, S,W-Co(OH)2/NF requires only an overpotential of 70 mV to achieve a current density of 10 mA cm-2 (vs. RHE). The HER performance of the dual-doped catalyst was optimized by tuning the W content and electrodeposition time, which led to improved catalytic activity. This was achieved by synthesizing S,Wx-Co(OH)2/NF with tungsten chloride-to-total metal ion molar ratios of x = 0.01, 0.05, and 0.1. As shown in [Supplementary Figure 6], the S,W0.05-Co(OH)2/NF electrode, with an intermediate W concentration, exhibits superior electrochemical performance compared to the other variants. In addition, LSV curves for S,W-Co(OH)2/NF deposited for 10, 20, and 30 min are presented in [Supplementary Figure 7]. Notably, the sample deposited for 30 min exhibits substantially improved HER performance compared to those prepared at shorter deposition times. The variation in charge transfer resistance (Rct) among different samples originates from differences in dopant concentration and structural disorder induced during electrodeposition. No additional crystalline phases were detected by XRD, and XPS confirms lattice incorporation rather than phase segregation. The optimized co-doped sample achieves a balance between defect generation and electronic conductivity, resulting in minimized Rct. Furthermore, the Tafel slope was used to evaluate reaction kinetics across different catalysts, providing insight into their electrocatalytic efficiency and mechanisms. As shown in [Figure 3B], the S,W-Co(OH)2/NF electrode exhibits the lowest Tafel slope (25 mV dec-1), which is significantly lower than those of S-Co(OH)2/NF (83 mV dec-1), W-Co(OH)2/NF (167 mV dec-1), and pristine Co(OH)2/NF (117 mV dec-1). This considerable drop indicates that the dual-doped catalyst allows for more efficient HER kinetics. Specifically, S,W-Co(OH)2/NF requires η = 70, 117, and 136 mV at j = 10, 50, and 100 mA cm-2, respectively (vs. RHE), as illustrated in [Figure 3C]. These outcomes are markedly inferior to those recorded for S-Co(OH)2/NF (134, 191, and 217 mV), W-Co(OH)2 (241, 305, and 334 mV), and Co(OH)2 (272, 366, and 419 mV). As summarized in [Supplementary Table 2], the dual-doped catalyst requires a lower overpotential at 10 mA cm-2 and exhibits faster HER kinetics than many previously reported catalysts. EIS provides additional evidence of the improved interfacial charge-transfer capability of the dual-doped catalyst. As presented in [Supplementary Figure 8], this catalyst shows the smallest charge transfer resistance (Rct = 7.4 Ω), markedly lower than those of S-Co(OH)2/NF (15.0 Ω), W-Co(OH)2/NF (60.1 Ω), and Co(OH)2/NF (28.0 Ω). These findings confirm that dual doping enhances electron transport efficacy at the electrolyte -electrode interface. Beyond intrinsic kinetics, the catalytic performance also benefits from an increased density of electrochemically accessible active sites. ECSA is an important parameter for evaluating catalyst performance. In this work, ECSA was estimated from Cdl, which was derived from CV measurements at various scan rates [Supplementary Figure 9]. S,W-Co(OH)2/NF electrode exhibits a Cdl of 111 mF cm-2, which is much higher than those of S-Co(OH)2/NF (61 mF cm-2), W-Co(OH)2/NF (4 mF cm-2), and Co(OH)2/NF (8 mF cm-2). This large Cdl value suggests that S,W-Co(OH)2/NF possesses more exposed electrochemically active sites, as shown in [Figure 3D]. To effectively compare the intrinsic HER activity across various catalysts, the current density was normalized to ECSA. The normalized LSV curves, as illustrated in [Supplementary Figure 10], show that S,W-Co(OH)2/NF exhibits superior HER activity compared to the other catalysts.
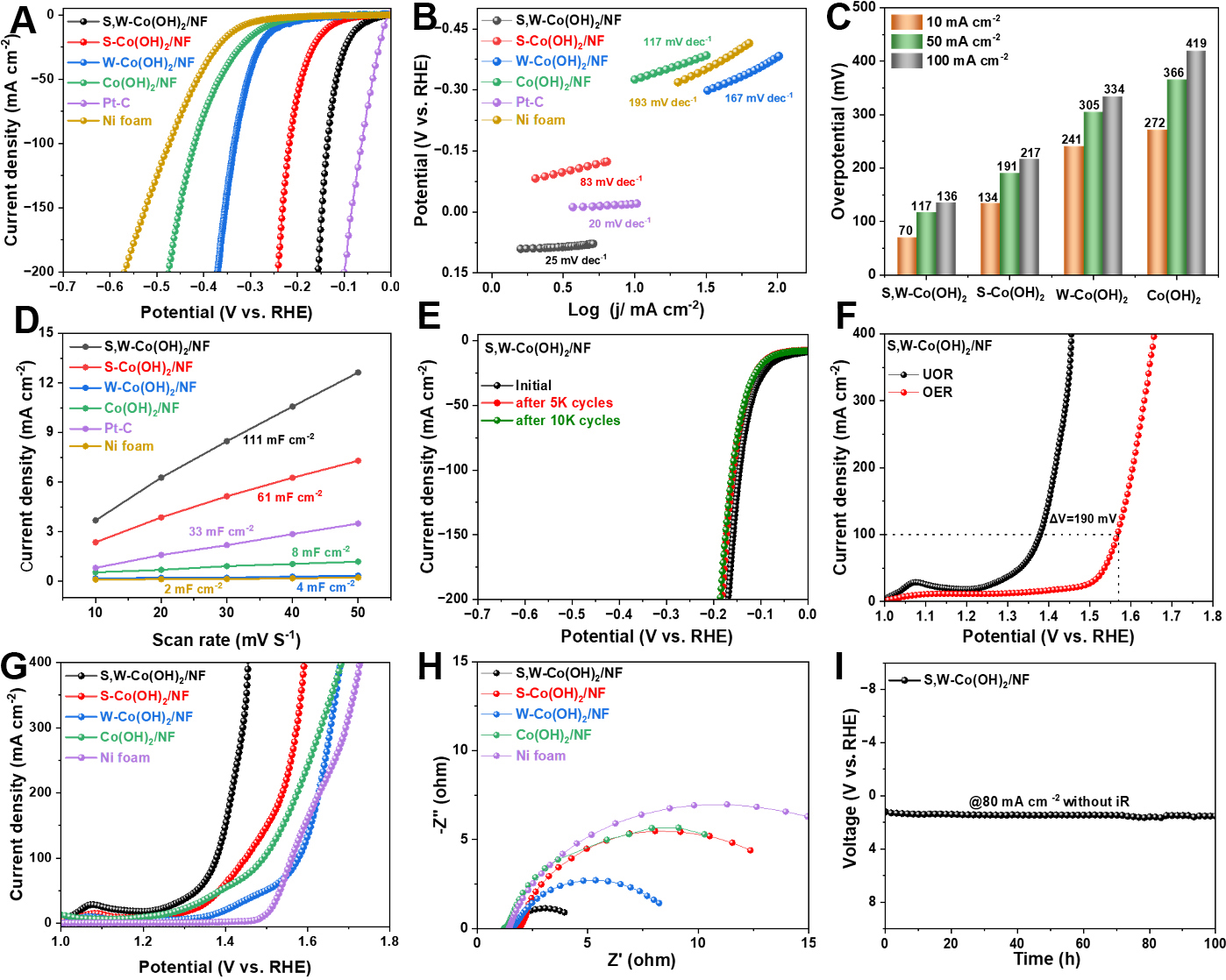
Figure 3. Electrochemical HER and UOR activity. (A) LSV curves and corresponding (B) Tafel slopes for HER; (C) Comparisons of overpotentials; (D) Calculated electrochemical Cdl; (E) LSV curves of S,W-Co(OH)2/NF before and after 5,000 and 10,000 CV cycles during HER testing; (F) LSV comparison of UOR and OER for S,W-Co(OH)2/NF; (G) LSV curves for UOR of the as-prepared catalysts (S,W-Co(OH)2/NF achieves 1.41 V at 200 mA cm-2 and 1.45 V at 500 mA cm-2); (H) EIS profiles of various samples recorded at 0.45 V during UOR testing; (I) UOR stability of S,W-Co(OH)2/NF. RHE: Reversible hydrogen electrode: NF: nickel foam; HER: hydrogen evolution reaction; UOR: urea oxidation reaction; LSV: linear sweep voltammetry; Cdl: double-layer capacitance; CV: cyclic voltammetry; OER: cyclic voltammetry; EIS: electrochemical impedance spectroscopy.
These findings indicate that W doping not only increases the number of active sites in S-Co(OH)2/NF but also significantly enhances its intrinsic HER activity. Moreover, long-term durability is critical for electrocatalysts in practical applications and is evaluated using various techniques. As shown in [Figure 3E and Supplementary Figure 11], the LSV curves and Nyquist plots of S,W-Co(OH)2/NF demonstrate no significant alterations after 5,000 and 10,000 cycles, indicating excellent stability. The variation in Nyquist plots after cycling can be attributed to surface reconstruction and electrochemical activation during prolonged operation. Such structural evolution is common in TMHs under alkaline conditions. Despite changes in interfacial resistance, the polarization curves remain nearly unchanged, indicating the formation of a stable and catalytically active surface phase. Furthermore, the voltage remains stable without noticeable degradation during chronoamperometric measurement at -80 mA cm-2 for approximately 100 h, as illustrated in [Supplementary Figure 12]. This outstanding performance underscores the potential usefulness of S,W-Co(OH)2/NF in hydrogen production.
Electrochemical UOR
The electrochemical performance of UOR was assessed in 1 M KOH containing 0.5 M urea using a standard three-electrode configuration. The deposition duration and W loading of S,W-Co(OH)2/NF were systematically optimized to enhance catalytic performance. Analysis of the LSV curves indicates that a deposition time of 30 min is optimal [Supplementary Figure 13]. Meanwhile, compositional optimization of S,Wx-Co(OH)2/NF (x = 0.01, 0.05, and 0.1) reveals that S,W0.05-Co(OH)2/NF shows the best catalytic activity and was therefore selected for further investigation [Supplementary Figure 14]. The S,W-Co(OH)2/NF catalyst achieves a current density of 100 mA cm-2 for UOR at 1.37 V vs. RHE, as depicted in [Figure 3F]. Notably, this potential is 190 mV lower than that required for OER at the same current density, indicating that UOR is thermodynamically more favorable than OER. Furthermore, as shown in [Figure 3G], S,W-Co(OH)2/NF achieves current densities of 200 and 500 mA cm-2 at potentials of 1.41 and 1.45 V vs. RHE, respectively. These findings demonstrate a clear reduction compared with single-doped and pristine catalysts. As illustrated in [Supplementary Figure 15], UOR reaches current densities of 10, 50, and 100 mA cm-2 at applied potentials of 1.02, 1.32, and 1.37 V vs. RHE, respectively. These results surpass the performance of most reported UOR catalysts, as detailed in [Supplementary Table 3]. For systematic comparison, the dual-doped catalyst was examined alongside the corresponding single-doped materials, the pristine catalyst, and the bare NF. As shown in [Supplementary Figure 16], kinetic analysis shows that S,W-Co(OH)2/NF exhibits a Tafel slope of only 13 mV dec-1, which is substantially lower than those of S-Co(OH)2/NF (66 mV dec-1), W-Co(OH)2/NF (21 mV dec-1), Co(OH)2/NF (45 mV dec-1), and bare NF (93 mV dec-1). These results indicate that the dual-doped catalyst delivers faster electrocatalytic kinetics toward UOR. In addition, the Rct of S,W-Co(OH)2/NF is significantly lower than that of the other catalysts, as presented in [Figure 3H], suggesting enhanced electrode kinetics for UOR. The LSV curves of the dual-doped catalyst were also recorded in 1 M KOH with different urea concentrations, as shown in [Supplementary Figure 17]. The results demonstrate that the current density increases with increasing urea concentration from 0.1 M to 1 M. To ensure sufficient urea availability during long-term stability testing, a concentration of 0.5 M urea in 1.0 M KOH was selected. Chronoamperometric measurements reveal that S,W-Co(OH)2/NF maintains a current density of 80 mA cm-2 over 100 h, as shown in [Figure 3I], confirming its excellent long-term stability for UOR. The ECSA of the catalysts was evaluated in a urea-containing electrolyte by estimating the Cdl from CV at different scan rates, as depicted in [Supplementary Figure 18]. S,W-Co(OH)2/NF demonstrates a Cdl value of 74 mF cm-2, which is significantly higher than those of S-Co(OH)2/NF (38 mF cm-2), W-Co(OH)2/NF (3 mF cm-2), and NF (2 mF cm-2), as shown in [Supplementary Figure 19].
Overall activity for water and urea splitting
Because of its excellent activity toward both HER and UOR, overall water and urea splitting devices were constructed using two identical dual-doped catalyst electrodes as the cathode and anode, respectively. In this section, “cell voltage” refers to measurements in a two-electrode configuration, whereas “potential vs. RHE” refers to three-electrode measurements. This setup is designed to assess practical applicability, as illustrated in [Figure 4A]. The HER LSV curves of S,W-Co(OH)2/NF remain nearly identical in the absence and presence of 0.5 M urea in the electrolyte, suggesting that urea has a negligible effect on HER performance [Supplementary Figure 20]. Notably, this dual-electrode system exhibits exceptional activity for overall urea-assisted electrolysis under alkaline conditions, as demonstrated in [Figure 4B]. For urea-assisted electrolysis, the assembled device requires only 1.19 V to reach 10 mA cm-2, whereas conventional water splitting requires 1.54 V under identical conditions. This performance is comparable to previously reported electrocatalysts for urea-aided water splitting, as summarized in [Supplementary Table 4]. Based on the cell voltage at 10 mA cm-2, the energy consumption of urea-assisted electrolysis is reduced by approximately 23% compared with conventional water electrolysis. The detailed calculation is provided in the Supporting Information.
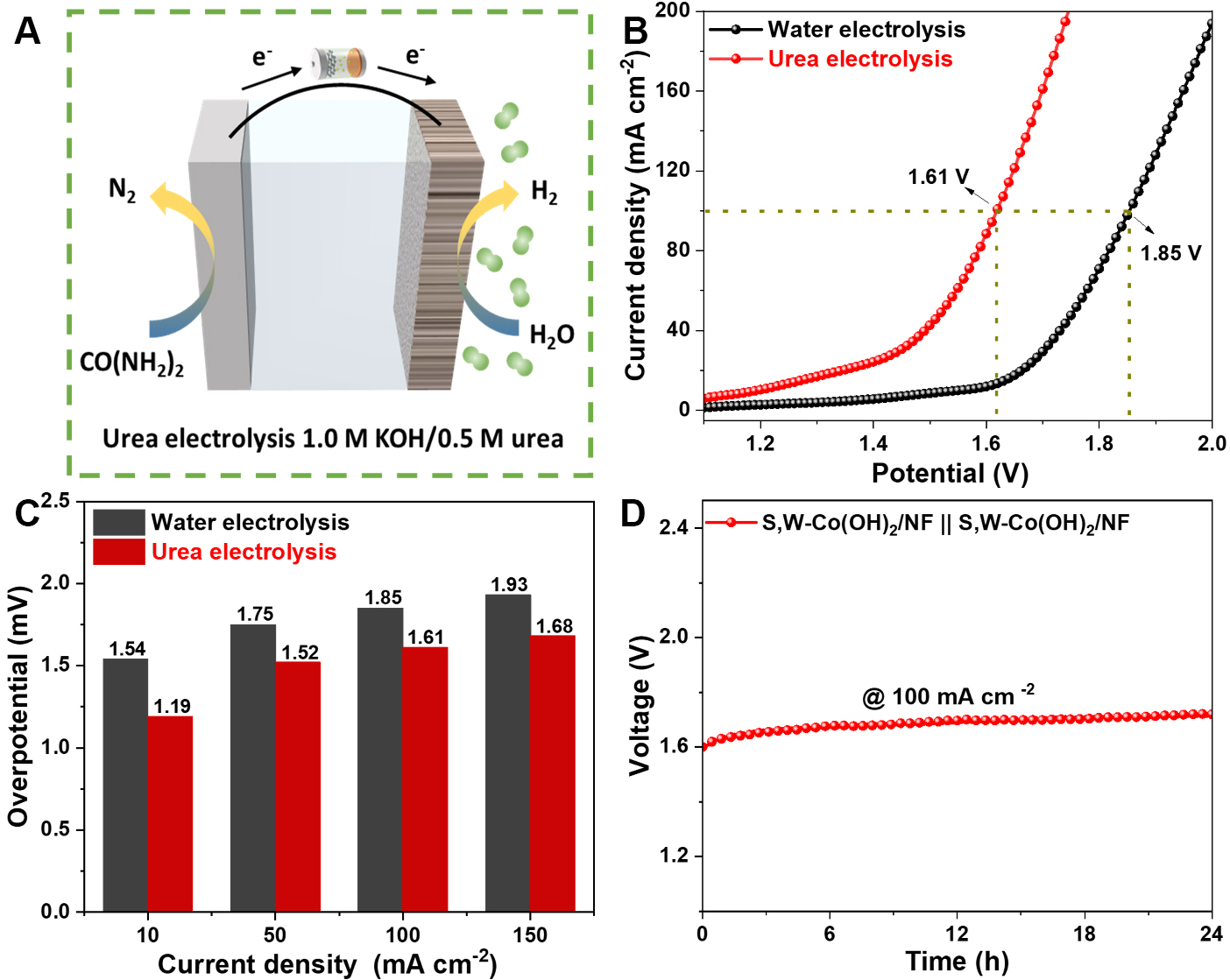
Figure 4. (A) Schematic of the two-electrode configuration employing S,W-Co(OH)2/NF; (B) LSV curves of two identical S,W-Co(OH)2/NF electrodes in 1.0 M KOH containing 0.5 M urea; (C) Comparison of cell voltages at 10, 50, 100, and 150 mA cm-2 for water and urea splitting; (D) Stability performance. NF: Nickel foam; LSV: linear sweep voltammetry.
To enable electrolysis at higher current densities of 50, 100, and 150 mA cm-2, as depicted in [Figure 4C], the required cell voltages for water decomposition are 1.75, 1.85, and 1.93 V, respectively. In contrast, the corresponding voltages for urea electrolysis are markedly reduced to 1.52, 1.61, and 1.68 V under the same conditions. This difference highlights the system’s potential for energy-efficient hydrogen generation and the utilization of urea-rich wastewater. While catalytic activity is important, stability over time is a critical factor for practical catalyst applications. Notably, the S,W-Co(OH)2/NF electrode demonstrates excellent stability, maintaining effective urea electrolysis over 24 h despite a significant reduction in current density, as illustrated in [Figure 4D]. These results emphasize the robust stability of the catalyst. Collectively, these findings indicate that the dual-functional S,W-Co(OH)2/NF displays exceptional catalytic efficacy in concurrent water and urea electrolysis, making it a promising candidate for hydrogen evolution systems.
Post-HER and post-UOR characterizations of the dual-doped catalyst were conducted using XRD, SEM, and XPS. The XRD pattern in [Supplementary Figure 21] confirms the presence of the Co(OH)2 phase. SEM images [Supplementary Figure 22] verify the electrode's exceptional endurance. The XPS data [Supplementary Figure 23] indicate enhanced chemical stability after electrolysis. These findings demonstrate outstanding structural and chemical stability, further highlighting the superior performance of S,W-Co(OH)2/NF for urea-assisted water splitting compared to current catalysts.
DFT calculations
To gain deeper insight into the intrinsic relationship between the electronic structure of the doped hydroxide system and its catalytic behavior, DFT calculations were performed on representative single-doped and dual-doped model structures, as illustrated in [Figure 5A-C]. Differential charge density maps [Supplementary Figure 24] reveal the complementary electronic effects of S and W dopants in Co(OH)2. S doping induces charge accumulation at adjacent Co sites and charge depletion around nearby O atoms, indicating electron donation that enhances Co-O covalency. In contrast, W doping causes electron depletion around the W-O unit and charge accumulation on neighboring Co atoms, reflecting electron withdrawal by high-valence W and the resulting local lattice polarization. The dual-doped S,W-Co(OH)2 [Figure 5D] integrates these effects, producing coupled charge accumulation-depletion regions and long-range polarization. This redistributes electron density by enriching certain Co sites while creating electron-deficient Co-O-W domains, a built-in electronic asymmetry that facilitates urea activation and oxygenation steps, thereby accelerating UOR kinetics[37]. The observed charge redistribution around Co sites after W and S co-doping modifies the local electronic structure, which directly influences adsorption energetics. For HER, the optimized electronic configuration adjusts the hydrogen adsorption free energy (ΔGH*) toward a more favorable value, thereby accelerating reaction kinetics. For UOR, the redistributed charge enhances interactions with reaction intermediates and facilitates electron transfer. These theoretical results are consistent with the experimentally observed reduced overpotential and lower charge transfer resistance. To further elucidate the effect of S and W incorporation on the electronic configuration, the projected density of states (PDOS) of the modeled structures was examined. The corresponding d-band centers relative to the Fermi level (EF) are calculated to be -0.258, -0.293, and -0.270 eV for the S-doped, W-doped, and dual-doped structures, respectively [Figure 5E and Supplementary Figure 25]. The S-doped sample exhibits the d-band center closest to EF, the W-doped sample the farthest, while the dual-doped sample lies in between, indicating a finely tuned intermediate adsorption strength consistent with its balanced catalytic behavior[17,38]. In the context of HER, it is well established that ΔGH∗ is the pivotal parameter, with the optimal value ideally approaching zero[39]. As depicted in [Figure 5F], S,W-Co(OH)2 delivers a near-thermoneutral ΔGH* of 0.37 eV, consistent with its fastest HER kinetics. In contrast, W-Co(OH)2 binds H∗ too weakly, while S-Co(OH)2 exhibits intermediate binding. These trends are consistent with the DCD/PDOS results: S doping enriches Co 3d states, while W induces electron withdrawal via W-O bonds; dual doping synergistically integrates these effects to achieve near-ideal H∗ adsorption. Free-energy profiles [Figure 5G] identify dehydrogenation as the rate-determining step (RDS). The highest energy barrier for S,W-Co(OH)2 is 1.80 eV, lower than those of S-Co(OH)2 (1.99 eV) and W-Co(OH)2 (1.91 eV). This reduction is linked to dual doping-induced electronic polarization; electron-rich Co sites promote urea activation/dehydrogenation, while electron-deficient Co-O-W domains stabilize oxygenated intermediates, collectively lowering the reaction barrier. The urea oxidation pathway and corresponding optimized configurations for the single and dual-doped models are shown in [Figure 5H and Supplementary Figure 26]. Overall, the combined experimental and theoretical findings demonstrate that synergistic anion-cation co-doping is an effective electronic structure modulation strategy for developing highly active bifunctional catalysts for water and urea splitting.
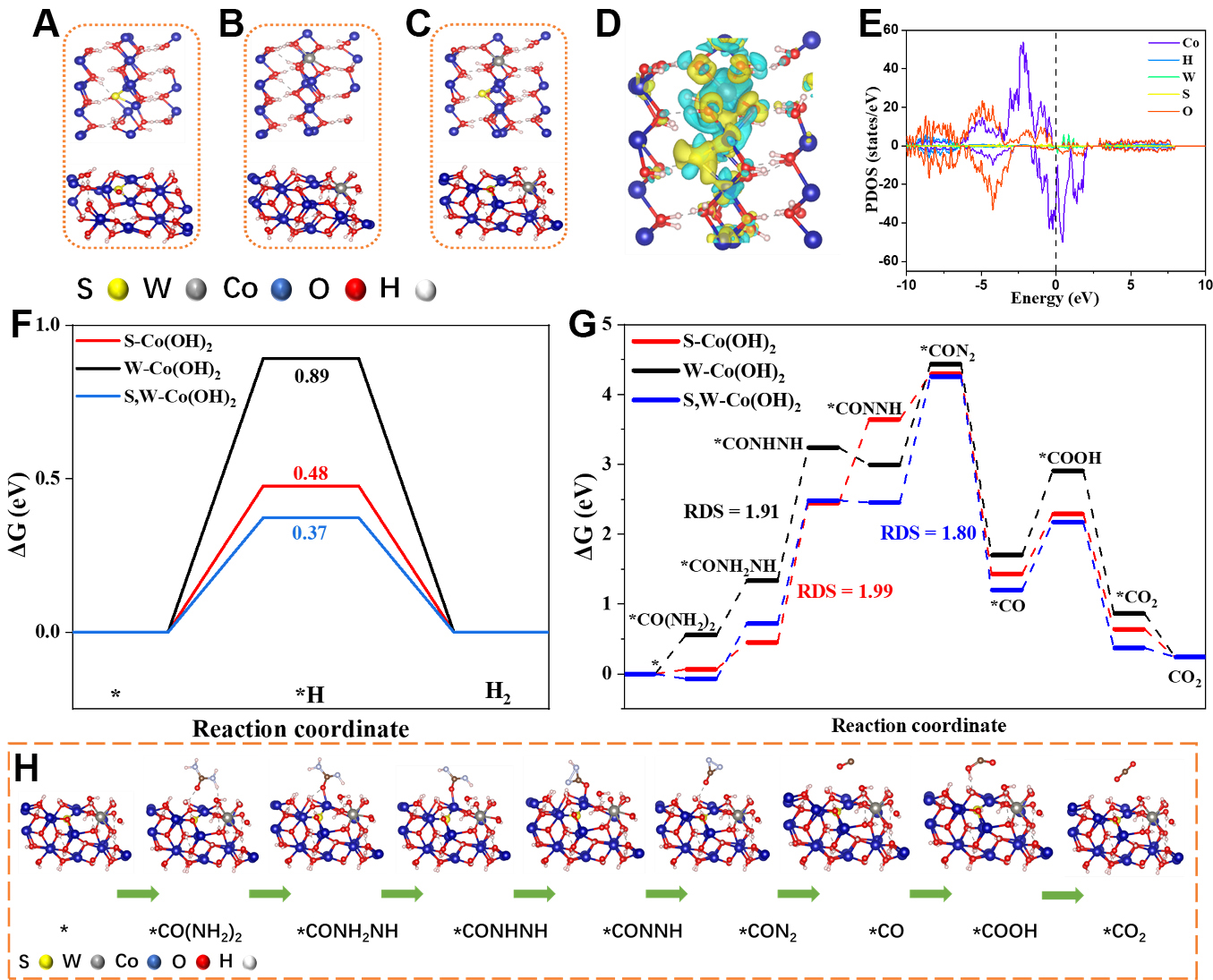
Figure 5. DFT analysis. (A-C) Optimized structural models of (A) S-Co(OH)2; (B) W-Co(OH)2; and (C) S,W-Co(OH)2; (D) Differential charge density of S,W-Co(OH)2. Yellow and cyan regions denote electron accumulation and depletion, respectively; (E) PDOS; (F) HER free-energy diagram; (G) UOR free-energy profiles on S-Co(OH)2, W-Co(OH)2, and S,W-Co(OH)2; (H) Diagrammatic representation of the urea oxidation mechanism and corresponding configurations. PDOS: Projected density of states; DFT: density functional theory; HER: hydrogen evolution reaction; UOR: urea oxidation reaction.
CONCLUSIONS
In this study, a co-doping approach involving an anionic dopant (S) and a cationic dopant (W) was employed to regulate the electronic structure of Co(OH)2 for enhanced hydrogen evolution and urea oxidation. The optimized S,W-Co(OH)2/NF catalyst exhibits excellent HER activity, requiring an overpotential of 70 mV to achieve a current density of 10 mA cm-2, and also shows strong UOR performance, delivering 500 mA cm-2 at 1.45 V vs. RHE, along with stable operation for 100 h. DFT analysis indicates that the simultaneous incorporation of S and W modifies the Co d-band center, leading to nearly thermoneutral hydrogen adsorption and a reduced energy barrier for urea dehydrogenation. These results highlight an effective strategy for designing bifunctional electrocatalysts and suggest a promising route for low-energy hydrogen generation integrated with wastewater treatment.
DECLARATIONS
Authors’ contributions
Conceptualization: Khan, S.
Methodology: Khan, S.
Sample preparation: Khan, S.; Feng, Y.
Electrochemical testing: Khan, S.
Data curation: Khan, S.
Software: Khan, S.
Formal analysis: Khan, S.; Cheng, M.; Ahmad, N.
Validation: Khan, S.; Cheng, M.
Data analysis: Khan, S.
Writing - original draft: Khan, S.
Review, and editing: Khan, S.
Data acquisition and analysis: Feng, Y.
Conducted density functional theory calculations: Chen, Y.
Mingyu Cheng: Formal analysis, Validation, and
Visualization: Cheng, M.
Sample characterization: Li, Y.
Project supervision: Zhang, G.
Experimental planning: Zhang, G.
Data analysis: Zhang, G.
Manuscript preparation: Zhang, G.
Availability of data and materials
The data supporting the findings of this study are available from the corresponding author upon reasonable request.
AI and AI-assisted tools statement
During the preparation of this manuscript, the author(s) used ChatGPT (version 3.0, released 2025-09-10) was used solely for language editing. The tool did not influence the study design, data collection, analysis, interpretation, or the scientific content of the work. All authors take full responsibility for the accuracy, integrity, and final content of the manuscript.
Conflicts of interest
Zhang, G. is an Associate Editor of the journal Micro Nano Science. He was not involved in any step of the editorial process, including reviewer selection, manuscript handling, or decision making. The other authors declared that there are no conflicts of interest.
Financial support and sponsorship
This work was supported by the National Key R&D Program of China (2022YFA1504001), the National Natural Science Foundation of China (T2325021, 22321001), and the CAS (Chinese Academy of Sciences) Project for Young Scientists in Basic Research (YSBR-070). The numerical calculations in this study were performed at the Supercomputing Center of the University of Science and Technology of China.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
Supplementary Materials
REFERENCES
1. Staffell, I.; Scamman, D.; Velazquez Abad, A.; et al. The role of hydrogen and fuel cells in the global energy system. Energy. Environ. Sci. 2019, 12, 463-91.
3. Roger, I.; Shipman, M. A.; Symes, M. D. Earth-abundant catalysts for electrochemical and photoelectrochemical water splitting. Nat. Rev. Chem. 2017, 1, 0003.
4. Seh, Z. W.; Kibsgaard, J.; Dickens, C. F.; Chorkendorff, I.; Nørskov, J. K.; Jaramillo, T. F. Combining theory and experiment in electrocatalysis: Insights into materials design. Science 2017, 355, eaad4998.
5. Seitz, L. C.; Dickens, C. F.; Nishio, K.; et al. A highly active and stable IrOx/SrIrO3 catalyst for the oxygen evolution reaction. Science 2016, 353, 1011-4.
6. Verma, S.; Kim, B.; Jhong, H. R. “.; Ma, S.; Kenis, P. J. A. A gross-margin model for defining technoeconomic benchmarks in the electroreduction of CO2. ChemSusChem 2016, 9, 1972-9.
7. Tolba, R.; Tian, M.; Wen, J.; Jiang, Z.; Chen, A. Electrochemical oxidation of lignin at IrO2-based oxide electrodes. J. Electroanal. Chem. 2010, 649, 9-15.
8. Zhang, Z.; Lin, F.; Xiang, L.; et al. Synergistic effect for simultaneously catalytic ozonation of chlorobenzene and NO over MnCoO catalysts: byproducts formation under practical conditions. Chem. Eng. J. 2022, 427, 130929.
9. Yu, Q.; Liu, X.; Liu, G.; et al. Constructing three-phase heterojunction with 1D/3D hierarchical structure as efficient trifunctional electrocatalyst in alkaline seawater. Adv. Funct. Mater. 2022, 32, 2205767.
10. Boggs, B. K.; King, R. L.; Botte, G. G. Urea electrolysis: direct hydrogen production from urine. Chem. Commun. 2009, 4859.
11. Zhan, G.; Hu, L.; Li, H.; et al. Highly selective urea electrooxidation coupled with efficient hydrogen evolution. Nat. Commun. 2024, 15, 5918.
12. Chen, W.; Xu, L.; Zhu, X.; et al. Unveiling the electrooxidation of urea: intramolecular coupling of the N-N bond. Angew. Chem. Int. Ed. 2021, 60, 7297-307.
13. Mccrory, C. C. L.; Jung, S.; Peters, J. C.; Jaramillo, T. F. Benchmarking heterogeneous electrocatalysts for the oxygen evolution reaction. J. Am. Chem. Soc. 2013, 135, 16977-87.
14. Carmo, M.; Fritz, D. L.; Mergel, J.; Stolten, D. A comprehensive review on PEM water electrolysis. Int. J. Hydrogen. Energy. 2013, 38, 4901-34.
15. Gao, X.; Zhang, S.; Wang, P.; Jaroniec, M.; Zheng, Y.; Qiao, S. Urea catalytic oxidation for energy and environmental applications. Chem. Soc. Rev. 2024, 53, 1552-91.
16. Zhang, X.; Feizpoor, S.; Humayun, M.; Wang, C. Urea oxidation reaction electrocatalysts: correlation of structure, activity, and selectivity. Chem. Catalysis. 2024, 4, 100840.
18. Nørskov, J. K.; Bligaard, T.; Logadottir, A.; et al. Trends in the exchange current for hydrogen evolution. J. Electrochem. Soc. 2005, 152, J23.
19. Subbaraman, R.; Tripkovic, D.; Strmcnik, D.; et al. Enhancing hydrogen evolution activity in water splitting by tailoring Li+-Ni(OH)2-Pt interfaces. Science 2011, 334, 1256-60.
20. Danilovic, N.; Subbaraman, R.; Strmcnik, D.; et al. Enhancing the alkaline hydrogen evolution reaction activity through the bifunctionality of Ni(OH)2/metal catalysts. Angew. Chem. Int. Ed. 2012, 51, 12495-8.
21. Medvedev, J. J.; Delva, N. H.; Klinkova, A. Mechanistic analysis of urea electrooxidation pathways: key to rational catalyst design. ChemPlusChem 2024, 89, e202300739.
22. Upadhyay, P.; Deka, A.; Chakma, S. Unlocking efficient electrochemical urea oxidation and understanding mechanism insights of Co-doped NiS. ACS. Eng. Au. 2025, 5, 450-67.
23. Yang, M.; Liu, Y.; Ge, W.; Liu, Z. Enhanced electrocatalytic activity of sulfur and tungsten co-doped nickel hydroxide nanosheets for urea oxidation. Colloid. Surface. A. 2023, 665, 131226.
24. Peng, K.; Liu, L.; Bhuvanendran, N.; et al. Effective regulation on catalytic performance of nickel-iron-vanadium layered double hydroxide for urea oxidation via sulfur incorporation. Mater. Adv. 2023, 4, 1354-62.
25. Liu, Y.; Yang, Z.; Zou, Y.; Wang, S.; He, J. Trace cobalt doping and defect engineering of high surface area α‐Ni(OH)2 for electrocatalytic urea oxidation. Energy. Environ. Mater. 2023, 7, e12576.
26. Fei, L.; Sun, H.; Xu, X.; et al. Understanding the bifunctional catalytic ability of electrocatalysts for oxygen evolution reaction and urea oxidation reaction: recent advances and perspectives. Chem. Eng. J. 2023, 471, 144660.
27. Chatenet, M.; Pollet, B. G.; Dekel, D. R.; et al. Water electrolysis: from textbook knowledge to the latest scientific strategies and industrial developments. Chem. Soc. Rev. 2022, 51, 4583-762.
28. Hafner, J. Ab‐initio simulations of materials using VASP: density‐functional theory and beyond. J. Comput. Chem. 2008, 29, 2044-78.
29. Perdew, J. P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865-8.
30. Liu, Y.; Koza, J. A.; Switzer, J. A. Conversion of electrodeposited Co(OH)2 to CoOOH and Co3O4, and comparison of their catalytic activity for the oxygen evolution reaction. Electrochim. Acta. 2014, 140, 359-65.
31. Sun, J.; Huang, Z.; Huang, T.; et al. Defect-rich porous CoS1.097/MoS2 hybrid microspheres as electrocatalysts for pH-universal hydrogen evolution. ACS. Appl. Energy. Mater. 2019, 2, 7504-11.
32. Feng, L.; Yu, G.; Wu, Y.; et al. High-index faceted Ni3S2 nanosheet arrays as highly active and ultrastable electrocatalysts for water splitting. J. Am. Chem. Soc. 2015, 137, 14023-6.
33. Wang, H.; Li, Z.; Hong, S.; et al. Tungstate intercalated NiFe layered double hydroxide enables long-term alkaline seawater oxidation. Small 2024, 20, 2311431.
34. Koushik, D.; Verhees, W. J. H.; Zhang, D.; et al. Atomic layer deposition enabled perovskite/PEDOT solar cells in a regular n-i-p architectural design. Adv. Mater. Inter. 2017, 4, 1700043.
35. Du, P.; Wen, Y.; Chiang, F.; et al. Corrosion engineering to synthesize ultrasmall and monodisperse alloy nanoparticles stabilized in ultrathin cobalt (oxy)hydroxide for enhanced electrocatalysis. ACS. Appl. Mater. Interfaces. 2019, 11, 14745-52.
36. Sun, H.; Lian, Y.; Yang, C.; et al. A hierarchical nickel-carbon structure templated by metal-organicframeworks for efficient overall water splitting. Energy. Environ. Sci. 2018, 11, 2363-71.
37. Fotso, L. E. K.; Mehmood, S.; Rani, J. V.; Dutta, J.; Pal, U. Advances and outlook of nickel-based MOFs-LDHs materials for energy conversion. Adv. Sustain. Syst. 2025, 9, e00483.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0









Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].