


Context-dependent roles of alpha-ketoglutarate in brain disorders: molecular mechanisms and translational opportunities
 0
0 Abstract
Brain disorders, including neurodegenerative, psychiatric and oncologic disorders, represent a major global public health challenge, yet their underlying pathogenic mechanisms remain incompletely understood. Accumulating evidence suggests that mitochondrial dysfunction, oxidative stress, impaired energy metabolism, and neurotransmitter imbalance contribute to the etiology of these disorders. Restoring mitochondrial and metabolic function emerges as a potential therapeutic strategy. α-ketoglutarate (AKG), a key intermediate in the tricarboxylic acid cycle, plays diverse roles in cellular energy metabolism, amino acid biosynthesis, redox regulation and epigenetic control. Preclinical studies indicate that exogenous AKG supplementation or targeted modulation of AKG-related metabolic pathways can influence mitochondrial homeostasis as well as cellular metabolic and epigenetic states. However, the biological effects of AKG appear to be context dependent, varying across disease states and metabolic conditions. This review synthesizes current evidence on the molecular mechanisms through which AKG regulates mitochondrial, metabolic, and epigenetic processes in the nervous system, highlighting its distinct and sometimes divergent roles across neurological conditions. By integrating findings from diverse disease contexts, this review aims to critically assess the therapeutic potential of targeting AKG-related pathways in brain disorders and to outline key challenges and priorities for future translational research.
Keywords
INTRODUCTION
Brain disorders, including neurodegenerative diseases, psychiatric conditions, and brain tumors, represent a substantial global public health burden and rank among the leading causes of mortality and long-term disability worldwide[1]. Despite significant advances in molecular and genetic research, effective therapeutic interventions for these disorders remain limited, underscoring the complexity and heterogeneity of the underlying disease etiologies. Convergent lines of evidence indicate that mitochondrial dysfunction, oxidative stress, and impaired energy metabolism constitute shared pathological hallmarks across diverse brain disorders. For example, mitochondrial DNA mutations and oxidative stress act synergistically to accelerate aging, which is the primary risk factor for neurodegenerative diseases[2]. As the central regulator of cellular energy metabolism, mitochondria not only generate adenosine triphosphate (ATP) but also regulate apoptosis[3,4], maintain calcium homeostasis[5], mediate thermogenesis[6], and support the biosynthesis of essential metabolites, including amino acids and cholesterol[7,8]. Beyond these metabolic functions, mitochondria play critical roles in neuronal development and migration, and in synaptic function[9]. Given the central pathological role of mitochondrial dysfunction in brain disorders, strategies targeting mitochondrial function may offer a promising avenue for therapeutic intervention.
The mitochondrial tricarboxylic acid (TCA) cycle is a core metabolic pathway underpinning oxidative phosphorylation and is integral to a wide range of biological processes including cellular energy production and biosynthesis. Beyond its canonical metabolic role, accumulating evidence demonstrates that TCA cycle metabolites actively regulate physiological processes and participate in disease pathogenesis[4]. As a key intermediate in the TCA cycle, α-ketoglutarate (AKG) is essential for ATP generation, but also functions as a pleiotropic metabolic regulator with anti-inflammatory, anti-oxidant, and anti-apoptotic properties
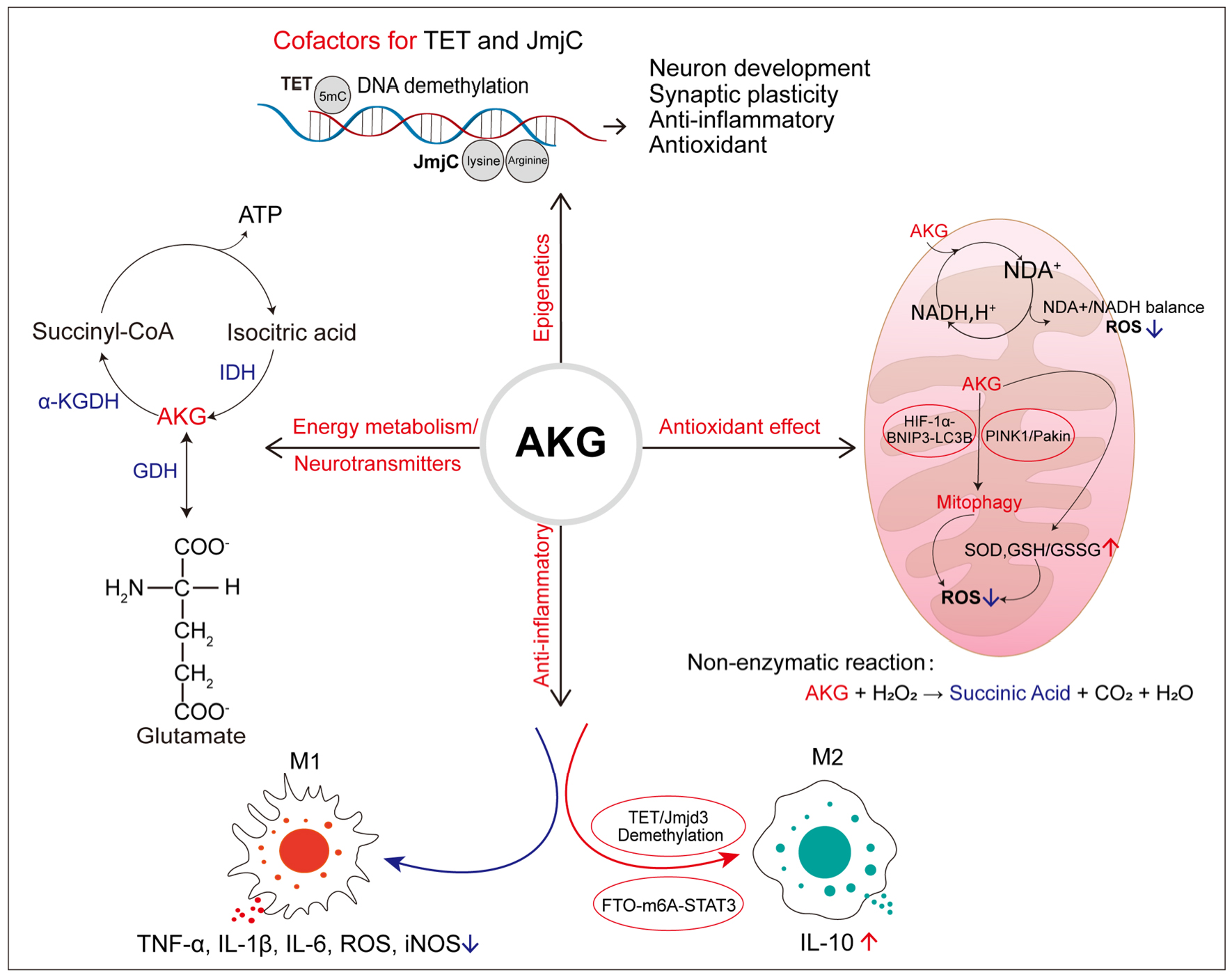
Figure 1. Major physiological functions of α-KGDH. AKG participates in the tricarboxylic acid (TCA) cycle and serves as a key node linking carbon and nitrogen metabolism. It exerts antioxidant effects enzymatically by maintaining the oxidized/reduced nicotinamide adenine nucleotide (NAD+/NADH) ratio, promoting mitophagy, and enhancing the intracellular antioxidant defense system, and via non-enzymatic mechanisms by scavenging hydrogen peroxide. Its anti-inflammatory action is mediated through the modulation of macrophage polarization (M1/M2). AKG acts as an essential cofactor for 2-oxoglutarate-dependent dioxygenases (2-OGDDs), including enzymes from the ten-eleven translocation (TET) and Jumonji C (JmjC) families, thereby regulating epigenetic processes via the modulation of DNA and histone methylation. α-KGDH: α-Ketoglutarate dehydrogenase;BNIP3: BCL2/adenovirus E1B 19-kDa-interacting protein 3; FTO-m6A: fat mass and obesity associated N⁶-methyladenosine deaminase; GDH: glutamate dehydrogenase; GSH: glutathione; GSSG: glutathione disulfide; HIF-1α: hypoxia-inducible factor 1-alpha; IDH: isocitrate dehydrogenase; IL-1β: interleukin-1 beta; IL-6: interleukin-6; IL-10: interleukin-10; iNOS: inducible nitric oxide synthase; LC3B: microtubule-associated protein 1 light chain 3 beta; PINK: PTEN-induced kinase 1; ROS: reactive oxygen species; SOD: superoxide dismutase; STAT3: signal transducer and activator of transcription 3; TNF-α: tumor necrosis factor alpha; 5mC: 5-methylcytosine.
PROPERTIES AND BIOLOGICAL ROLES OF AKG
Biochemical properties of AKG
AKG, also known as 2-ketoglutarate or α-ketoglutamic acid, is a water-soluble, weak organic acid with the molecular formula C5H6O5 (HOOC-CH2-CO-CH2-COOH). In experimental research, AKG is most commonly administered in its salt forms, such as calcium or sodium salts[13,14], or as lipophilic derivatives[11]. These salt forms do not substantially alter intestinal absorption efficiency of AKG, which occurs primarily in the upper small intestine[15,16]. Furthermore, animal studies have shown that AKG-derived metabolites can be detected in the cerebrospinal fluid, providing indirect evidence that AKG or its metabolic products are capable of crossing the blood-brain barrier[15].
Roles in energy metabolism and neurotransmitter biosynthesis pathways
AKG occupies a central position within the TCA cycle. It is generated from isocitrate via an oxidative decarboxylation reaction catalyzed by isocitrate dehydrogenase (IDH) and is subsequently converted to succinyl-CoA through a second oxidative decarboxylation reaction catalyzed by α-ketoglutarate dehydrogenase (α-KGDH)[17], thereby providing the foundation for mitochondrial ATP production. Beyond its canonical role in oxidative metabolism, AKG also serves as a metabolic hub linking carbon and nitrogen metabolism, contributing to glutamate synthesis and nitrogen excretion. In mitochondria, glutamate dehydrogenase (GDH) catalyzes the reversible interconversion of AKG and glutamate through reductive amination or oxidative deamination, directly coupling carbon flux to nitrogen handling[18,19]. This reaction is tightly regulated by the cellular energy state: GDH activity is stimulated by adenosine diphosphate (ADP) and inhibited by ATP, establishing a feedback mechanism that coordinates oxidized/reduced nicotinamide adenine dinucleotide (NAD+/NADH) availability with amino-acid metabolism[20,21].
Additionally, AKG serves as an amino-group acceptor in reactions catalyzed by alanine aminotransferase and aspartate aminotransferase, leading to the biosynthesis of glutamate and other non-essential amino acids. AKG can be converted to glutamate through transamination[22], and subsequently to gamma-aminobutyric acid (GABA) through decarboxylation. Given that glutamate and GABA serve as the principal excitatory and inhibitory neurotransmitters, respectively, AKG plays an important role in maintaining the balance of neuronal excitability. Under glucose deprivation, exogenous AKG supplementation increases intracellular glutamate levels and inhibits cancer cell death driven by overexpression of the cystine/glutamate antiporter solute carrier family 7 member 11 (SLC7A11) and glutamate depletion[23]. Collectively, these findings underscore the pivotal role of AKG in bridging mitochondrial energy metabolism and neurotransmitter synthesis.
Antioxidant function
Excessive production of reactive oxygen species (ROS) is a major pathogenic driver in brain disorders, where it directly compromises mitochondrial integrity and function, ultimately leading to energy failure, synaptic degeneration, and neuronal loss[24-26]. In this context, AKG has been shown to exert significant antioxidant effects. In the TCA cycle, AKG serves as a substrate for α-KGDH, donating a hydride ion to NAD+, which is reduced to NADH. As the primary electron donor for the mitochondrial respiratory chain, NADH directly participates in the generation of ROS by influencing electron transport chain activity and the NAD+/NADH ratio[27-29]. Through this mechanism, AKG acts as a metabolic mediator that helps regulate cellular redox homeostasis. In addition, AKG mitigates oxidative stress by regulating mitochondrial quality control processes. Specifically, AKG enhances mitophagy through the activation of the PTEN-induced kinase 1 (PINK1)/parkin pathway and the hypoxia-inducible factor 1-alpha (HIF-1α)-BCL2/adenovirus E1B 19-kDa-interacting protein 3 (BNIP3) microtubule-associated protein 1 light chain 3 beta (LC3B) axis, promoting the removal of damaged mitochondria. This targeted clearance reduces excessive ROS production and alleviates oxidative stress-induced cellular damage[11,30,31].
AKG exerts its antioxidant function through both enzymatic and non-enzymatic mechanisms. At the level of endogenous antioxidant defense, AKG upregulates the activity of superoxide dismutase (SOD) by activating the constitutive androstane receptor (CAR) signaling pathway and the sirtuin 1 (SIRT1)/peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α)/nuclear factor erythroid 2-related factor 2 (Nrf2) pathway, thereby promoting the transcriptional regulation of antioxidant enzymes[32,33]. In parallel, AKG alleviates oxidative damage by reducing malondialdehyde accumulation, increasing the ratio of reduced to oxidized glutathione (GSH/glutathione disulfide (GSSG)), and modulating the ETS variant transcription factor 4 (ETV4)/SLC7A11/glutathione peroxidase 4 (GPX4) axis to suppress lipid peroxidation and excessive ROS accumulation[34]. In addition to these enzyme-mediated effects, AKG functions as a direct ROS scavenger by participating in a non-enzymatic decarboxylation reaction with hydrogen peroxide, yielding succinate, water, and carbon dioxide[35]. Consistent with this mechanism, studies in Drosophila melanogaster have demonstrated that AKG effectively mitigates toxicity induced by sodium nitroprusside and hydrogen peroxide through the non-enzymatic antioxidant mechanisms[36].
Anti-inflammatory role
Immune cells, including macrophages and microglia, exhibit functional plasticity and can polarize toward either pro-inflammatory (M1) or anti-inflammatory (M2) phenotypes. In the central nervous system, oxidative stress induced by ROS accumulation activates innate immune signaling pathways and initiates a self-perpetuating cycle of oxidative stress and inflammation. Excessive accumulation of ROS activates the mitogen-activated protein kinase (MAPK), nuclear factor κB (NF-κB), and NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3) inflammasome signaling pathway[37-39], driving microglia toward a pro-inflammatory M1-like state and promoting the release of pro-inflammatory cytokines, thereby sustaining neuroimmune activation. In turn, inflammatory mediators produced by activated microglia further promote the generation of ROS and induce the expression of inducible nitric oxide synthase[40,41], exacerbating the oxidative stress and ultimately causing neuronal injury and apoptosis.
AKG plays an important regulatory role in the process of macrophage polarization. Experimental evidence shows that AKG significantly increases the M2/M1 macrophage ratio through a ten-eleven translocation (TET) enzyme-mediated DNA demethylation mechanism[42]. In parallel, AKG has been shown to drive M2 polarization via activation of the fat mass and obesity-associated (FTO) N6-methyladenosine deaminase (m6A)-signal transducer and activator of transcription 3 (STAT3) signaling pathway[43]. Moreover, as a key downstream metabolite of glutamine, AKG accumulation promotes M2 macrophage polarization and exerts anti-inflammatory effects via multiple complementary mechanisms. These include the enhancement of DNA demethylation in a Jumonji domain-containing protein D3 (Jmjd3)-dependent manner[44,45], as well as the inhibition of sirtuin 5 (SIRT5)-mediated deacetylation of pyruvate dehydrogenase[46]. Under conditions of immune-inflammatory dysregulation, AKG further suppresses inflammatory responses by directly reducing the levels of multiple pro-inflammatory factors, including IL-6, IL-1β, and tumor necrosis factor-alpha (TNF-α)[12,47,48], as well as IL-4, IL-25, IL-3, and thymic stromal lymphopoietin (TSLP)[49], further highlighting its broad anti-inflammatory effects.
Roles in epigenetic modification
AKG functions as a key regulator at the intersection of cellular metabolism and epigenetic modification, serving as an essential co-substrate for 2-oxoglutarate-dependent dioxygenases (2-OGDDs). This enzyme family plays a central role in epigenetic and transcriptional regulation, and includes the TET family of DNA demethylase enzymes[50] and Jumonji C (JmjC) domain-containing histone demethylases, both of which catalyze hydroxylation reactions critical for chromatin remodeling[51]. Through TETmediated oxidation, 5-methylcytosine (5mC) is converted to 5-hydroxymethylcytosine (5-hmC), dynamically modulating the DNA methylation states that regulate early embryonic growth and development[52] and the maintenance of stem cell pluripotency[53]. Beyond its role in embryogenesis, 5-hmC-mediated epigenetic modification is critical in postnatal neurodevelopment and aging[54]. Notably, neurons possess high levels of 5-hmC that further increase in response to neural activity, supporting transcriptional plasticity required for learning and memory. In neurons, GDH, which catalyzes the conversion of glutamate to AKG, has a stimulatory effect on TET3 demethylation activity. Neuronal activation increases the levels of AKG, supporting its role in epigenetic regulation during neural plasticity[55]. Conversely, reduced AKG levels compromise TET3-dependent demethylation, thereby facilitating aberrant epigenetic states that promote tumor cell proliferation[56]. Collectively, these findings highlight AKG as a versatile metabolic regulator that exerts broad biological effects through epigenetic modulation, particularly in neural function, development, and disease.
AKG AND NEURODEGENERATIVE DISEASES
Core pathological features of major neurodegenerative diseases
Neurodegenerative diseases (NDDs) such as Alzheimer's disease (AD) and Parkinson's disease (PD) share pathological features centered on mitochondrial dysfunction and impaired energy metabolism. These metabolic alterations are accompanied by increased oxidative stress, chronic neuroinflammation, and progressive neuronal loss, collectively driving neurodegeneration[57]. The neuroanatomical patterns across NDDs are nevertheless disease-specific. In AD, cytochrome c oxidase activity is reduced by approximately 30%-50% in cortical and hippocampal regions, and presynaptic mitochondria exhibit early reductions in number along with swelling and cristae disorganization[58,59]. In contrast, in PD, mitochondrial complex I activity is impaired in neurons of the substantia nigra pars compacta[60]. Converging evidence from studies in animal models, postmortem human brain tissue analyses, and metabolic imaging studies indicates that these abnormalities precede overt clinical manifestations, with reduced glucose uptake and oxidative metabolism in detected vulnerable brain regions during prodromal stages[61,62].
Within this context of compromised energy metabolism, oxidative stress and neuroinflammation act as mutually reinforcing processes to accelerate disease progression. In NDDs, elevated ROS levels are usually associated with activation of microglia and astrocytes[40]. Excessive ROS not only causes direct neuronal damage, but also potentiates microglial activation and pro-inflammatory cytokine release, thereby sustaining a neuroinflammatory environment and exacerbating neurodegeneration[63]. Diseasespecific proteinopathies further intensify these pathogenic cascades: in AD, amyloid beta aggregation aggravates oxidative and inflammatory stress, whereas in PD, α-synuclein aggregation disrupts mitochondrial function and perturbs calcium homeostasis[64,65].
Chronic oxidative stress and inflammation disrupt mitochondrial bioenergetics as well as the dynamic balance between mitochondrial fission and fusion. These changes lead to alterations in mitochondrial morphology, impair mitochondrial delivery to presynaptic terminals, and ultimately reduce local ATP supply and Ca2+ buffering capacity[66]. The disruptions also impede the clearance or repair of damaged mitochondria, leading to the accumulation of dysfunctional organelles[67], which elevates ROS production and triggers inflammatory responses, thereby accelerating neurodegeneration[68]. Collectively, disruptions in energy metabolism, redox homeostasis imbalance, and sustained neuroinflammation represent pathological features shared across NDDs. The convergence of these mechanisms establishes a strong biological rationale for targeting metabolic intermediates AKG, thereby supporting further investigation of AKG-related pathways in preclinical and translational studies of NDDs.
AKG and Alzheimer’s disease
In AD, existing evidence linking AKG to disease mechanisms can be broadly categorized into direct intervention studies and indirect metabolic observations. Direct preclinical evidence has primarily implicated AKG in the regulation of synaptic plasticity and autophagy-related pathways. In the APP/PS1 mouse model of AD, administration of AKG or calcium α-ketoglutarate (Ca-AKG) ameliorated impaired long-term potentiation at hippocampal CA1 synapses, with more pronounced effects observed in female AD mice. Moreover, Ca-AKG facilitated synaptic tagging and capture, a well-established cellular readout for associative memory. These effects were mediated by N-methyl-D-aspartate receptor (NMDAR)‐independent but L‐type calcium channel-dependent signaling, as pharmacological inhibition of L-type Ca2+ channels or Ca2+-permeable α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors abolished the effects of Ca-AKG treatment[69].
Notably, Ca-AKG also upregulated the expression of autophagy markers, producing a rescue phenotype comparable to that achieved with rapamycin treatment in APP/PS1 AD mice[69]. Rapamycin is known to upregulate autophagy by inhibiting mammalian target of rapamycin (mTOR), a key regulator of translation initiation and long‐term synaptic plasticity, and has been shown to improve synaptic and mitochondrial function, ameliorate cognitive deficits, and reduce amyloid burden[70,71]. Consistent with these observations, AKG has been associated with lifespan extension in multiple model organisms, including mice, Caenorhabditis elegans, and Drosophila, potentially through inhibition of ATP synthase and mTOR signaling and induction of autophagy[12,72,73]. Together, these findings suggest that AKG may modulate autophagy-related signaling pathways, possibly involving mTOR signaling, although direct mechanistic evidence in AD models remains limited. Importantly, the regulatory relationship between AKG and mTOR appears to be bidirectional. On one hand, AKG may activate mTOR by replenishing the TCA cycle intermediate pools[74]. On the other hand, rapamycin treatment restores reduced AKG and glutamate levels in NADH:ubiquinone oxidoreductase subunit S4 (NDUFS4)-deficient models of mitochondrial encephalopathy, linking mTOR inhibition to the recovery of TCA-associated metabolites[75]. These observations raise the possibility that AKG influences the mTOR–autophagy axis beyond its role as a canonical TCA-cycle intermediate. Nevertheless, this view is supported mainly by preclinical models and indirect metabolic analyses, and the relevance of AKG modulation to human AD pathophysiology remains uncertain.
Indirect metabolic evidence further supports an association between AKG-related metabolic changes and impaired glucose oxidation in AD. In AD brains, dysfunctional insulin signaling is associated with reduced pyruvate dehydrogenase activity, which may constrain TCA cycle flux and limit the production of downstream metabolites, including AKG[76]. In male 3xTg-AD mice, dietary ketone ester supplementation partially restored several TCA cycle intermediates, and hippocampal AKG levels showed a strong positive correlation with glutamate concentrations (R = 0.852)[77]. While these findings reflect a concomitant restoration of TCA cycle and amino acid pools, they do not provide direct evidence for carbon flux from AKG into neurotransmitter synthesis. Overall, the existing evidence remains model-specific and largely indirect, leaving the causal contribution of AKG to AD pathogenesis unresolved.
AKG and Parkinson’s disease
PD is characterized by progressive degeneration of nigrostriatal dopaminergic neurons and the pathological accumulation of α-synuclein. Mitochondrial dysfunction and oxidative stress are widely recognized as contributors to PD pathophysiology, with numerous studies implicating deficits in mitochondrial complex I. In the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) mouse model of PD, MPTP is metabolized to 1-methyl-4-phenylpyridinium (MPP+), a potent inhibitor of mitochondrial complex I that triggers acute bioenergetic failure and oxidative stress in the nigrostriatal dopaminergic system[78,79]. In this MPTP mouse model, AKG administration improved motor coordination to a degree comparable to that observed with Madopar treatment and attenuated parameters of oxidative stress and mitochondrial complex I dysfunction. Specifically, AKG increased glutathione (GSH) levels and SOD activity while reducing malondialdehyde content, consistent with restored redox homeostasis and partial recovery of complex I function. AKG treatment also reduced α-synuclein accumulation and mitigated the loss of tyrosine hydroxylase-positive neurons in the midbrain, indicating a protective effect on dopaminergic neuronal integrity[80]. Collectively, these findings support a neuroprotective role for AKG in toxin-induced models of PD, but because these models primarily capture acute mitochondrial toxicity rather than the full spectrum of PD pathogenesis, these results do not establish therapeutic efficacy across distinct PD subtypes or disease stages.
To date, evidence derived from α-synuclein-driven PD models remains limited and awaits independent replication. In adeno-associated virus-loaded human α-synuclein mice and in A53T α-synuclein transgenic mice, dietary Ca-AKG supplementation has been reported to improve motor performance and attenuate dopaminergic neuron loss in the substantia nigra[81]. These functional and neuroprotective effects were accompanied by increased microglial expression of complement component C1q, an elevated LC3-II/LC3-I ratio indicative of enhanced autophagy activity, and reduced levels of phosphorylated α-synuclein. Notably, although C1q has been implicated in aberrant synaptic pruning and neuronal injury[82], it also contributes to the clearance of misfolded proteins and mitigation of neurotoxicity[83,84]. Accordingly, Ca-AKG may facilitate microglial autophagic degradation and reduce pathological α-synuclein burden. However, whether C1q is mechanistically required for the observed effects of AKG in α-synuclein-driven PD models remains unresolved and warrants further investigation.
AKG-related effects have also been reported in other neurodegenerative and neuroinjury-associated disorders characterized by bioenergetic impairment and mitochondrial dysfunction; however, the overall evidence base remains sparse. Among these disorders, ischemic stroke currently has the most direct support for AKG involvement, including early clinical exploration of ornithine AKG in stroke patients[85] and subsequent experimental studies demonstrating that AKG attenuates cerebral ischemia-reperfusion injury[86]. In contrast, evidence in intracerebral hemorrhage is largely indirect and primarily centers on epigenetic alterations associated with AKG-dependent signaling pathways[87], whereas the most direct interventional data in hemorrhagic brain injury derive from subarachnoid hemorrhage models rather than intracerebral hemorrhage itself[88]. In amyotrophic lateral sclerosis (ALS), the clearest intervention-related signal comes from metabolic therapies containing arginine AKG[89], complemented by additional studies reporting disruptions in TCA cycle intermediates and associated metabolic pathways[90]. By comparison, direct AKG supplementation studies in Huntington’s disease are currently lacking, with existing evidence pointing to disruption of the AKG metabolic axis[91] and AKG-dependent dioxygenase pathways[92]. Overall, these observations suggest that AKG may be implicated across a broader spectrum of neurodegenerative and brain injury-related disorders; however, supporting evidence remains limited and largely indirect.
AKG AND PSYCHIATRIC DISORDERS
Core metabolic and neurochemical abnormalities
Schizophrenia (SCZ), bipolar disorder (BD), and depressive disorder are major psychiatric conditions characterized by disturbances in cognition, mood, and behavior. Although these disorders differ in clinical manifestations and diagnostic criteria, they share several pathological features. For example, neuroimaging studies have identified overlapping patterns of brain structural abnormalities across multiple psychiatric disorders, although these abnormalities are usually subtle[93,94]. In addition, resting-state functional magnetic resonance imaging analyses have consistently revealed default mode network dysfunction and aberrant large-scale network interactions across diverse psychiatric conditions[95,96].
In addition to neural network dysfunctions, imbalances in neurotransmitter systems are also thought to underlie the etiology of psychiatric disorders. In SCZ, subcortical dopaminergic hyperactivity is associated with positive symptoms, whereas reduced cortical dopaminergic activity is linked to negative symptoms and cognitive impairment[97,98]. Gluck et al.[99] measured the activities of nine enzymes involved in glutamate and GABA metabolism in the postmortem dorsolateral prefrontal cortex from aged individuals with SCZ and matched controls. Their analysis revealed a two-fold increase in glutamic acid decarboxylase activity and a four-fold increase in phosphate-activated glutaminase activity in SCZ patients, indicating substantial disruption of glutamate and GABA metabolism. In BD, fluctuations in mood state parallel cyclical alterations in dopaminergic transmission[100]. In depressive disorder, reduced serotonergic and altered noradrenergic signaling in corticolimbic circuits have been implicated in anhedonia and abnormal stress responsiveness[101]. Taken together, these observations support a pathological framework centered on disrupted interactions between neural networks and neurotransmitter systems across major psychiatric disorders.
Mitochondria serve as a critical interface between energy metabolism pathways and neurotransmitter metabolism. Beyond their core function in ATP production, mitochondria regulate cellular redox homeostasis, autophagy, and calcium signaling pathways; consequently, mitochondrial dysfunction can disrupt neurotransmitter release, synaptic plasticity, and network oscillatory rhythms[102]. Mitochondrial dysfunction has emerged as a common pathological feature across major psychiatric disorders[103]. A meta-analysis of [18F]fluorodeoxyglucose positron electron tomography studies revealed hypometabolism in the frontal cortex of individuals with SCZ[104]. Consistent with these observations, mitochondrial complex I deficiency and altered activities of key TCA cycle enzymes were found in postmortem analyses of SCZ brains compared to controls[105,106]. In BD, manic episodes are typically associated with hypermetabolic patterns, whereas depressive episodes more closely resemble hypometabolic states with reduced oxidative phosphorylation efficiency[100]. Because neurotransmitter synthesis and cycling depend on mitochondrial ATP supply[107], impaired oxidative metabolism and reduced ATP availability can lead to neurotransmitter imbalance implicated in psychiatric disorders.
AKG and schizophrenia
In SCZ, the evidence linking AKG to disease-related biology appears relatively more convergent than in other psychiatric disorders. A systematic review by Davison et al.[108] consistently identified reductions in TCA cycle-derived organic acids, including AKG, across multiple SCZ cohorts, suggesting a relatively stable downregulation of TCA cycle-related metabolism in SCZ patients. In a complementary study, Xuan et al.[109] performed a serum metabolomic analysis using gas chromatography–mass spectrometry in unmedicated SCZ patients and matched healthy controls. This analysis identified 22 discriminative metabolites, among which citrate and AKG levels were significantly reduced in the SCZ group[109]. In first-episode neuroleptic-naive SCZ patients, changes in urinary AKG from baseline to a 6-week risperidone monotherapy were correlated with changes in the Positive and Negative Syndrome Scale (PANSS) score[110], supporting a potential biomarker-level association between AKG metabolism and symptom improvement. Furthermore, another metabolomic study reported significantly decreased plasma AKG and malate concentrations in SCZ patients at medium and high risk for antipsychotic-induced metabolic syndrome supporting an association between metabolic syndrome risk and reduced levels of peripheral TCA cycle intermediates[111].
In addition to peripheral metabolite profiling, Bubber et al.[106] examined the TCA cycle enzyme activities in postmortem dorsolateral prefrontal cortex of SCZ patients and found reduced activities of aconitase, α-KGDH and succinate thiokinase, and increased activities of succinate dehydrogenase and malate dehydrogenase. α-KGDH catalyzes the conversion of AKG to succinyl-CoA with concomitant production of NADH[17]. Accordingly, reduced α-KGDH activity implies a dysregulation of AKG availability and perturbation of downstream TCA cycle-associated pathways in SCZ.
Direct mechanistic evidence linking AKG metabolism to SCZ related phenotypes was provided by
AKG and bipolar disorder
BD is characterized by recurrent shifts between manic and depressive mood states. Metabolomics analyses of serum samples from BD patients have revealed abnormalities in the citric acid cycle, the urea cycle, and amino acid metabolism[113]. Metabolic profiles in BD vary across the manic, depressive, and mixed states, with particularly pronounced alterations in pathways governing the energy metabolism and amino acid utilization[114]. According to the "metabolic overdrive" hypothesis of BD, the manic state may be characterized by increased cerebral glycolysis and glutaminolysis, reflecting elevated energy demand and enhanced utilization of glycolytic and glutamatergic substrates[100]. Given that AKG occupies a central position in the TCA cycle and serves as a metabolic link between the glutamate–glutamine cycle and mitochondrial energy production, it is plausibly involved in the coupling of energy and amino acid metabolism in BD. Supporting a potential association, Yoshimi et al.[113] reported significantly elevated serum levels of AKG in BD patients compared with healthy controls. Nevertheless, current evidence remains largely indirect and does not yet establish a definitive role for AKG in BD pathophysiology.
Chronic lithium treatment, an established mood stabilizer, significantly increased serum AKG levels in rats. Since lithium is known to enhance mitochondrial respiratory chain activity, increase ATP production and reduce oxidative stress[115], this lithium-induced elevation of AKG underscores the potential of AKG as a biomarker for monitoring therapeutic efficacy. However, more evidence is still needed to support this implication. First, only male participants were enrolled in Yoshimi et al.’s study[113], necessitating larger, sex-inclusive cohorts of medication-free patients to clarify the relationship between AKG levels and mood state trajectories in BD. Second, it remains unclear whether interventions directly targeting AKG-related pathways can improve BD-associated phenotypes. Until these questions are addressed through more comprehensive and mechanistic studies, the association between AKG and BD remains uncertain.
AKG and depressive disorders
Reduced levels of brain-derived neurotrophic factor (BDNF) and impairment of the BDNF-mediated signaling have been implicated in the pathophysiology of depressive disorders. Direct evidence linking AKG to depression-related phenotypes has been derived primarily from preclinical studies, with the chronic social defeat stress (CSDS) model being particularly informative. In this model, depression-like behaviors are associated with a regional BDNF imbalance, characterized by decreased BDNF expression in the hippocampus and increased expression in the nucleus accumbens. AKG pretreatment normalized this BDNF imbalance by restoring hippocampal BDNF expression while restraining BDNF upregulation in the nucleus accumbens. These molecular effects were accompanied by alleviated social avoidance behavior in mice[116].
An open-label pilot study with a limited sample size examined metabolomic changes in depressive disorder patients treated with the traditional Chinese medicine formula Xiaoyaosan. Treatment was associated with a reduction in Hamilton Depression Rating Scale scores, accompanied by increased urinary levels of several endogenous metabolites, including creatinine, taurine, AKG, and xanthurenic acid, alongside decreased levels of energy-related metabolites such as citrate and lactate[117]. Although these findings suggest that AKG-related metabolic alterations may accompany symptomatic improvement, they do not establish AKG as a causal mediator of antidepressant effects. The available evidence further indicates that the behavioral effects of AKG may be influenced by the metabolic context. For example, long-term dietary AKG supplementation exacerbated anxiety-like behaviors in female mice fed with an obesogenic diet. Under these conditions, AKG supplementation also decreased the exploratory behavior, and was associated with increased expression of autophagy-related markers and decreased antioxidant enzyme activities in the cerebral cortex[118]. The anxiogenic effects observed in the context of an obesogenic diet may reflect a direct involvement of AKG in the metabolism of neurotransmitters such as glutamate and γ-aminobutyric acid as well as a reduced activity of mechanistic target-of-rapamycin complex 1 (mTORC1). Although anxiety-like behavior was the primary readout in this study, these findings remain informative for depressive disorders because anxiety and depressive phenotypes often overlap under conditions of chronic stress, implicating a potential association between AKG and depressive disorders.
AKG AND BRAIN TUMORS
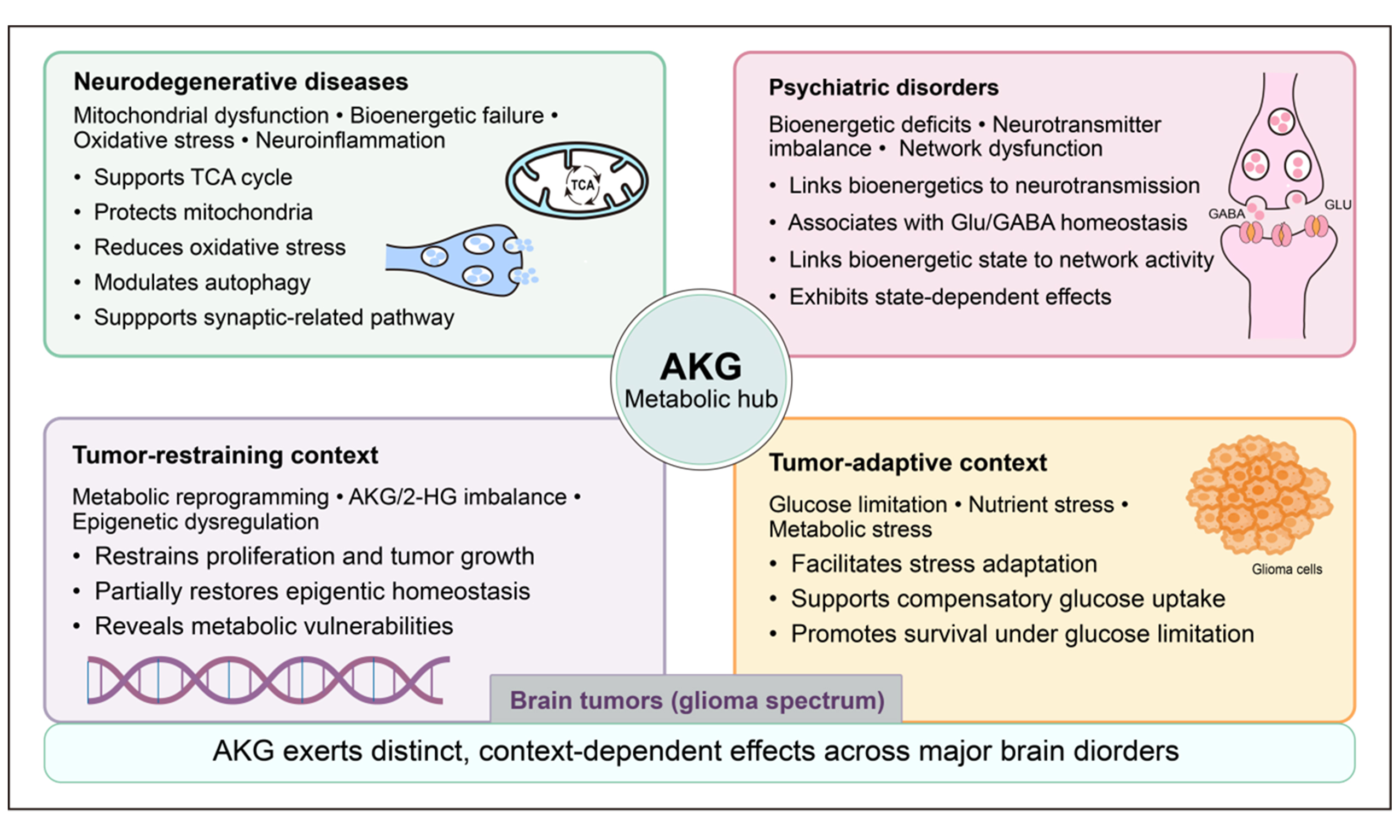
In contrast to neurodegenerative and major psychiatric disorders, brain tumors are primarily characterized by aberrant proliferation and profound metabolic reprogramming of tumor cells. Despite these fundamental pathological differences, accumulating basic and translational studies implicate AKG in gliomas development and progression. This section focuses on gliomas as a representative brain tumor entity, outlining the role of AKG in tumor-associated metabolic reprogramming and highlighting its context-dependent biological effects.
Core metabolic features of malignant gliomas
Gliomas, particularly glioblastoma, are the most common primary malignant intracranial tumors and are associated with an extremely poor prognosis, with 5-year survival rates below 7%[119]. A prominent pathological feature of glioblastoma is metabolic reprogramming, whereby tumor cells remain highly reliant on glycolysis even under normoxic conditions. In this context, a substantial amount of glucose is converted to lactate and exported from the tumor cell under aerobic conditions, a phenomenon known as the Warburg effect. Beyond ATP production, glycolysis and its associated branching pathways generate intermediates for the biosynthesis of nucleotides, fatty acids, and non-essential amino acids, thereby supporting sustained tumor cell proliferation. Despite elevated aerobic glycolysis, glioblastoma cells preserve functional mitochondrial metabolism and remain dependent on continuous replenishment of TCA cycle intermediates[120,121]. Under conditions of limited glucose availability, tumor cells shift carbon utilization toward alternative substrates such as glutamine to maintain the TCA cycle[122]. Glutamine is first converted to glutamate by glutaminase, and glutamate is subsequently transformed into AKG via glutamate dehydrogenase 1 (GDH1) or aminotransferases. This process provides anaplerotic input into the TCA cycle, thereby supporting oxidative phosphorylation and biosynthetic demands. Dysregulated glutamate uptake and release by glioma cells have been associated with peritumoral neural dysfunction and excitotoxicity highlighting the broader consequences of altered amino acid metabolism in the glioblastoma microenvironment[123].
Isocitrate dehydrogenase 1 and 2 (IDH1/2) are key metabolic enzymes that catalyze the oxidative decarboxylation of isocitrate to AKG. Mutations in IDH1/2 lead to aberrant conversion of AKG to the oncometabolite 2-hydroxyglutarate (2-HG)[124]. Due to its close structural similarity to AKG, 2-HG competitively inhibits multiple AKG-dependent dioxygenases, including the Jumonji domain–containing histone demethylases and TET family DNA demethylases, thereby inducing widespread DNA and histone hypermethylation and driving epigenetic dysregulation[125].
From an AKG-centric perspective, glioma pathology can be conceptualized along three interconnected axes: (1) metabolic reprogramming characterized by the Warburg effect and reliance on glutamine; (2) a glutamate–TCA cycle metabolic axis, with AKG serving as a central metabolic hub; and (3) IDH mutation–driven perturbation of the AKG/2-HG balance, leading to epigenetic reprogramming.
AKG in malignant gliomas
In gliomas, the effects of AKG are highly context-dependent, varying according to IDH mutation status, amino acid metabolism, and nutrient availability. Rather than exerting a uniform antitumor effect, AKG can either contribute to epigenetic and metabolic reprogramming or facilitate tumor adaptation under metabolic stress. In IDH–wild-type glioma, the expression of the gene encoding branched amino acid transaminase 1 (BCAT1), the enzyme that initiates the catabolism of branched-chain amino acids, is frequently upregulated and supports mitochondrial respiration and anabolic metabolism[126,127]. In U251MG and other IDH–wild-type glioblastoma cell lines, Zhang et al.[127] showed that BCAT1 knockout increased the NAD+/NADH ratio but impaired mitochondrial respiration, mTORC1 signaling, and nucleotide biosynthesis. Under these metabolically compromised conditions, supplementation with dimethyl-AKG (DMKG), a cell-permeable derivative of AKG, further exacerbated mitochondrial dysfunction, leading to depletion of ATP, nucleotides, and proteins, ultimately inducing tumor cell death[127]. Consistent with these findings, combined treatment with the BCAT1 inhibitor gabapentin and AKG resulted in synthetic lethality and suppressed tumor growth[127]. Taken together, these findings identify the BCAT1–AKG axis as a metabolic synthetic-lethal vulnerability in glioblastoma and support further investigation as a target for preclinical intervention studies.
In diffuse midline gliomas, including diffuse intrinsic pontine glioma (DIPG), histone H3.3K27M-related mutations are highly prevalent and are associated with aberrant chromatin accessibility and transcriptional profiles[128]. In this context, treatment with DMKG suppresses cell proliferation and neurosphere-forming capacity while inducing G1/S cell-cycle arrest. Transcriptomic and epigenomic profiling showed that DMKG downregulated genes associated with cell-cycle progression and DNA replication, while upregulating genes related to cellular stress-response and differentiation. At the level of histone modifications, DMKG treatment reduced H3K27 acetylation with only modest effects on H3K27 methylation, indicating that its biological effects are mediated predominantly through CREB-binding protein (CBP)/p300-dependent histone acetylation networks[129]. In contrast, IDH1/2-mutant gliomas are characterized by reduced AKG levels and accumulation of its antagonist 2-HG. 2-HG competitively inhibits AKG–dependent dioxygenases, leading to genome-wide histone and DNA methylation alterations[130]. IDH1 mutations, in particular, induce extensive DNA hypermethylation and alterations of histone modifications, reshaping the methylome in a manner that mirrors the changes observed in CpG island methylator phenotype-positive lower-grade gliomas[131]. In IDH-mutant glioma models, AKG supplementation or mutant pharmacologic inhibition of mutant IDH partially restores TET dioxygenase activities and improves DNA repair capacity[132]. Collectively, these findings support the therapeutic potential of AKG supplementation or modulation of the 2-HG/AKG balance as a means to relieve dioxygenase inhibition and mitigate epigenetic dysregulation in IDH-mutant gliomas.
Notably, AKG supplementation can, under specific metabolic conditions, promote glioma cell survival rather than suppress tumor growth. In glioblastoma models subjected to glucose limitation, GDH1 becomes phosphorylated at serine 384 and forms a complex with RelA and inhibitor of nuclear factor kappa-B kinase subunit beta (IKKβ). GDH1-derived AKG directly engages and activates the IKKβ-NF-κB signaling axis, thereby upregulating the expression of the glucose transporter 1 (GLUT1) to promote compensatory glucose uptake and tumor cell survival under low glucose conditions[133]. Consequently, disrupting the interaction between AKG and IKKβ inhibits tumor growth, whereas pharmacologic inhibition of NF-κB reduces tumor cell viability under low glucose in vitro[133].
FUTURE PERSPECTIVES
Table 1 summarizes the evidence for the core biochemical roles and the effects of AKG in brain disorders. Despite growing interest in AKG biology and the accumulating evidence for its roles in brain physiology and disease, several key mechanistic and translational questions remain unresolved. Different AKG formulations, including Ca-AKG, DMKG, and related derivatives, may exert distinct biological effects across experimental systems[134,135]. Accordingly, future studies should systematically compare these formulations under dose-equivalent conditions and rigorously define their pharmacokinetic and pharmacodynamic properties, including systemic exposure, transport across the blood–brain barrier, and effective delivery to brain tissue. The mechanism of AKG entry into the brain, its regional and cell-type-specific distribution, and its intracellular metabolic fate under physiological conditions remain poorly characterized. In this context, stable-isotope tracing approaches will be particularly valuable for clarifying how AKG is routed through the TCA cycle and neurotransmitter-linked metabolic pathways in the brain[136].
Representative evidence of the roles and effects of AKG across major brain conditions, grouped by evidence category
| Disease | Reference | Model/Cohort | AKG exposure/status | Main AKG-related findings |
| Panel A. Direct AKG supplementation/intervention | ||||
| AD | Navakkode et al., 2025[69] | Hippocampal slices from APP/PS1 mice | 1 mM Ca-AKG | Rescues long-term potentiation deficits; facilitates synaptic tagging and capture; increases LC3-II levels |
| PD | Satpute et al., 2013[80] | MPTP-induced PD mice | 500 mg/kg AKG | Improves motor performance; lowers oxidative stress; restores mitochondrial complex I activity; protects dopaminergic neurons; decreases midbrain α-synuclein levels |
| PD | Zhang et al., 2023[81] | AAV-human α-synuclein and A53T α-synuclein PD models | 2% Ca-AKG | Improves motor performance; reduces total α-synuclein and pSer129-α-synuclein; attenuates nigrostriatal dopaminergic neuron loss; increases microglial C1q; increases LC3 levels |
| SCZ | Zhang et al., 2023[112] | Human forebrain organoids | 10 μg/mL AKG | Partly restores TCA intermediates and GABA levels; normalizes aberrant network firing |
| Depressive disorders | Eid et al., 2025[116] | Adult male C57BL/6J mice | 300 mg/kg AKG | Reduces depressive-like behaviors and improves learning; normalizes BDNF levels in the hippocampus and nucleus accumbens |
| Depressive disorders | Demianchuk et al., 2025[118] | C57BL/6J mice on standard or cafeteria diet | 1% AKG | Under a cafeteria diet, increases anxiety-like behaviors; reduces cortical antioxidant enzyme activities; upregulates autophagy-related genes |
| Glioma spectrum | Zhang et al., 2022[127] | IDH-wild-type GBM cells | 10 mM DMKG | Further impairs oxidative phosphorylation, mTORC1 signaling; and nucleotide synthesis; depletes ATP; induces tumor cell death |
| Glioma spectrum | Lee et al., 2023[129] | DIPG cell lines (SF8628 and DIPG-007) | In vitro DMKG treatment | Reduces proliferation and neurosphere growth; induces G1/S arrest; suppresses H3K27 acetylation |
| Glioma spectrum | Wang et al., 2019[133] | U87 and U251 GBM cells | 0.5 mM methyl-α-KG | Activates IKKβ–NF-κB signaling; increases GLUT1 expression and glucose uptake; promotes GBM survival under glucose restriction |
| Panel B. Indirect AKG pathway modulation | ||||
| AD | Pawlosky et al., 2020[77] | Aged 3xTg-AD mice on a ketone-ester diet | Ketone-ester diet (no AKG treatment) | Partly restores hippocampal AKG, citrate and other TCA intermediates and amino acids; AKG shows a strong correlation with glutamate; behavioral improvement parallels TCA–glutamate remodeling |
| BD | Yoshimi et al., 2016[113] | BD patients vs controls; male rats treated with lithium or valproate | Blood metabolomics in BD; chronic mood-stabilizer treatment in rats with serum metabolomics (no AKG treatment) | BD serum shows TCA (citrate, AKG), urea-cycle and amino-acid abnormalities; lithium, but not valproate, increases serum AKG and selected TCA intermediates in rats |
| Depressive disorders | Tian et al., 2014[117] | Depressed patients treated with Xiaoyaosan | Urinary metabolomics (no AKG treatment) | Symptom improvement is accompanied by increased urinary AKG, creatinine, taurine, xanthurenic acid, and reduced citrate and lactate |
| Panel C. Biomarker level/associative evidence | ||||
| SCZ | Xuan et al., 2011[109] | First-episode, drug-naïve SCZ vs controls; serum metabolomics | Baseline serum TCA intermediates (no AKG treatment) | Reduced serum citrate and AKG, indicating lower circulating TCA-cycle intermediates |
| SCZ | Cai et al., 2012[110] | First-episode, drug-naïve SCZ on risperidone | Metabolomics during monotherapy | Longitudinal changes in urinary AKG, citrate and pregnanediol correlate with PANSS improvement, indicating TCA-related metabolic signatures of response |
| SCZ | Paredes et al., 2014[111] | Stable SCZ on SGAs, stratified by MetS risk vs controls | Plasma targeted metabolomics (no AKG treatment) | High/intermediate MetS-risk groups exhibited lower AKG and malate and higher glutamate, lipids and inflammatory markers, consistent with impaired AKG conversion and TCA intermediate depletion |
Large cohorts will also be required to determine whether alterations in AKG-related metabolic pathways are consistently observed across diagnostic groups, disease stages, treatment exposure, sex, and metabolic status. Any future AKG-based therapeutic intervention should be tailored to disease context and progression stage[69,81]. For example, in early neurodegenerative disorders, AKG may be best viewed as a metabolic support strategy, whereas in psychiatric disorders its relevance may be more strongly influenced by episode state, systemic metabolic background, and neurotransmitter homeostasis[99,106,114,137]. Similarly, in genetically stratified gliomas, patients should be subclassified according to their genetic background and tumor metabolic environment to guide rational AKG-based strategies[133].
Finally, issues related to safety, dose comparability, and biomarker development warrant careful consideration. Potential biomarkers include plasma or cerebrospinal fluid AKG-related metabolites, as well as measures of mitochondrial function, redox status, neurotransmitter homeostasis, and disease-relevant imaging or electrophysiological readouts.
CONCLUSIONS
Across neurodegenerative disorders, major psychiatric illnesses and glioblastoma, AKG should not be regarded as a uniformly protective factor. Instead, its biological roles are highly context-dependent. Across these pathological settings, AKG participates in central metabolic and regulatory processes, including the TCA cycle, mitochondrial function, autophagy and redox homeostasis, and modulates the epigenetic landscape through AKG-dependent dioxygenase activities. In models of neurodegenerative diseases and selected psychiatric disorders, AKG-linked pathways are predominantly associated with maintaining the TCA cycle flux, reinforcing antioxidant defenses and partially preserving synaptic integrity and neural network activity. By contrast, malignant gliomas may exploit AKG via BCAT1- and GDH1-dependent routes as a metabolic intermediate to support biosynthesis demands and redox balance, thereby enhancing tumor tolerance of nutrient and oxidative stress. Notably, most of the current evidence underlying these conclusions stems from cell lines, brain organoids and rodent models, with clinical evidence largely restricted to metabolomic associations and indirect markers of energy metabolism. Randomized controlled trials of AKG supplementation or targeted modulation of AKG-related pathways in neurological or oncological settings are still lacking. Overall, AKG is best conceptualized as a central metabolic hub linking energy metabolism, neurotransmitter homeostasis and epigenetic regulation in a disease state-dependent manner. Determining the conditions under which this metabolic hub can be safely and effectively exploited across distinct disease contexts remains a major challenge for future translational research.
DECLARATION
Acknowledgments
The graphical abstract was created by the authors using Adobe Illustrator (Adobe Inc.).
Authors’ contributions
Conducted the literature review and data curation, and initially drafted the manuscript: Xia Y, Wang H
Supervised the study and revised the manuscript: Meng Q
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
During the preparation of this manuscript, ChatGPT (OpenAI) was used solely for language polishing and readability improvement. The tool did not influence the study design, data collection, analysis, interpretation, or the scientific content of the work. All authors take full responsibility for the accuracy, integrity, and final content of the manuscript.
Financial support and sponsorship
This work was partially supported by grants from the National Natural Science Foundation of China (No. 82171504), the Natural Science Foundation of Hunan Province (No. 2022JJ20035), and the Science and Technology Innovation Program of Hunan Province (No. 2022RC1214).
Conflicts of Interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Steinmetz JD, Seeher KM, Schiess N, et al. Global, regional, and national burden of disorders affecting the nervous system, 1990-2021: a systematic analysis for the Global Burden of Disease Study 2021. Lancet Neurol. 2024;23:344-81.
2. Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787-95.
3. Woldetsadik AD, Vogel MC, Rabeh WM, Magzoub M. Hexokinase II-derived cell‐penetrating peptide targets mitochondria and triggers apoptosis in cancer cells. FASEB J. 2017;31:2168-84.
4. Martínez-reyes I, Chandel NS. Mitochondrial TCA cycle metabolites control physiology and disease. Nat Commun. 2020;11:102.
5. Olivas-Aguirre M, Torres-López L, Valle-Reyes JS, Hernández-Cruz A, Pottosin I, Dobrovinskaya O. Cannabidiol directly targets mitochondria and disturbs calcium homeostasis in acute lymphoblastic leukemia. Cell Death Dis. 2019;10:779.
6. Park A, Kim K, Park I, et al. Mitochondrial matrix protein LETMD1 maintains thermogenic capacity of brown adipose tissue in male mice. Nat Commun. 2023;14:3746.
7. Fang W, Jiang L, Zhu Y, et al. Methionine restriction constrains lipoylation and activates mitochondria for nitrogenic synthesis of amino acids. Nat Commun. 2023;14:2504.
8. Zhou Y, Wei J, Deng G, et al. Delivery of low-density lipoprotein from endocytic carriers to mitochondria supports steroidogenesis. Nat Cell Biol. 2023;25:937-49.
9. Tripathi K, Ben-shachar D. Mitochondria in the central nervous system in health and disease: the puzzle of the therapeutic potential of mitochondrial transplantation. Cells. 2024;13:410.
10. Gyanwali B, Lim ZX, Soh J, et al. Alpha-Ketoglutarate dietary supplementation to improve health in humans. Trends Endocrinol Metab. 2022;33:136-46.
11. Liu L, Zhang W, Liu T, et al. The physiological metabolite α-ketoglutarate ameliorates osteoarthritis by regulating mitophagy and oxidative stress. Redox Biol. 2023;62:102663.
12. Asadi Shahmirzadi A, Edgar D, Liao C, et al. Alpha-ketoglutarate, an endogenous metabolite, extends lifespan and compresses morbidity in aging mice. Cell Metab. 2020;32:447-56.e6.
13. Si X, Song Z, Liu N, Jia H, Liu H, Wu Z. α-Ketoglutarate restores intestinal barrier function through promoting intestinal stem cells-mediated epithelial regeneration in colitis. J Agric Food Chem. 2022;70:13882-92.
14. Sandalova E, Goh J, Lim ZX, et al. Alpha-ketoglutarate supplementation and BiologicaL agE in middle-aged adults (ABLE)—intervention study protocol. GeroScience. 2023;45:2897-907.
15. Filip R, Pierzynowski SG. The absorption, tissue distribution and excretion of enteraly administered α‐ketoglutarate in rats. J Anim Physiol Anim Nutr. 2007;92:182-9.
16. Lambert BD, Stoll B, Niinikoski H, Burrin DG, Pierzynowski S. Net portal absorption of enterally fed α-ketoglutarate is limited in young pigs. J Nutr. 2002;132:3383-6.
17. Hansen GE, Gibson GE. The α-ketoglutarate dehydrogenase complex as a Hub of plasticity in neurodegeneration and regeneration. Int J Mol Sci. 2022;23:12403.
18. Zaganas I, Spanaki C, Plaitakis A. Expression of human GLUD2 glutamate dehydrogenase in human tissues: Functional implications. Neurochem Int. 2012;61:455-62.
19. Plaitakis A, Kalef-Ezra E, Kotzamani D, Zaganas I, Spanaki C. The glutamate dehydrogenase pathway and its roles in cell and tissue biology in health and disease. Biology. 2017;6:11.
20. Herrero-Yraola A. Regulation of glutamate dehydrogenase by reversible ADP-ribosylation in mitochondria. EMBO J. 2001;20:2404-12.
21. Nassar OM, Wong KY, Lynch GC, Smith TJ, Pettitt BM. Allosteric discrimination at the NADH/ADP regulatory site of glutamate dehydrogenase. Protein Sci. 2019;28:2080-8.
22. Takeda K, Ishida A, Takahashi K, Ueda T. Synaptic vesicles are capable of synthesizing the VGLUT substrate glutamate from α‐ketoglutarate for vesicular loading. J Neurochem. 2012;121:184-96.
23. Koppula P, Zhang Y, Shi J, Li W, Gan B. The glutamate/cystine antiporter SLC7A11/xCT enhances cancer cell dependency on glucose by exporting glutamate. J Biol Chem. 2017;292:14240-9.
24. Hidalgo C, Arias-Cavieres A. Calcium, reactive oxygen species, and synaptic plasticity. Physiology. 2016;31:201-15.
25. Nissanka N, Moraes CT. Mitochondrial DNA damage and reactive oxygen species in neurodegenerative disease. FEBS Lett. 2018;592:728-42.
26. Wen P, Sun Z, Gou F, et al. Oxidative stress and mitochondrial impairment: key drivers in neurodegenerative disorders. Ageing Res Rev. 2025;104:102667.
27. Kussmaul L, Hirst J. The mechanism of superoxide production by NADH:ubiquinone oxidoreductase (complex I) from bovine heart mitochondria. Proc Natl Acad Sci USA. 2006;103:7607-12.
28. Ying W. NAD+/NADH and NADP+/NADPH in cellular functions and cell death: regulation and biological consequences. Antioxid Redox Signal. 2008;10:179-206.
29. Luo L, He Y, Zhao Y, et al. Regulation of mitochondrial NAD pool via NAD+ transporter 2 is essential for matrix NADH homeostasis and ROS production in Arabidopsis. Sci China Life Sci. 2019;62:991-1002.
30. An D, Zeng Q, Zhang P, et al. Alpha-ketoglutarate ameliorates pressure overload-induced chronic cardiac dysfunction in mice. Redox Biol. 2021;46:102088.
31. Yu H, Gan D, Luo Z, et al. α-Ketoglutarate improves cardiac insufficiency through NAD+-SIRT1 signaling-mediated mitophagy and ferroptosis in pressure overload-induced mice. Mol Med. 2024;30:15.
32. He L, Wu J, Tang W, et al. Prevention of oxidative stress by α-ketoglutarate via activation of CAR signaling and modulation of the expression of key antioxidant-associated targets in vivo and in vitro. J Agric Food Chem. 2018;66:11273-83.
33. Zhang Y, Wang H, Zhang Y, et al. α-Ketoglutarate ameliorates sarcopenia in D-galactose-induced aging mice by modulating protein homeostasis and optimizing mitochondrial function. Nutrients. 2025;17:3336.
34. He R, Wei Y, Peng Z, et al. α-Ketoglutarate alleviates osteoarthritis by inhibiting ferroptosis via the ETV4/SLC7A11/GPX4 signaling pathway. Cell Mol Biol Lett. 2024;29:88.
35. Liu S, He L, Yao K. The antioxidative function of alpha-ketoglutarate and its applications. BioMed Res Int. 2018;2018:1-6.
36. Bayliak MM, Shmihel HV, Lylyk MP, et al. Alpha-ketoglutarate attenuates toxic effects of sodium nitroprusside and hydrogen peroxide in Drosophila melanogaster. Environ Toxicol Pharmacol. 2015;40:650-9.
37. Liu Z, Yao X, Jiang W, et al. Advanced oxidation protein products induce microglia-mediated neuroinflammation via MAPKs-NF-κB signaling pathway and pyroptosis after secondary spinal cord injury. J Neuroinflammation. 2020;17:90.
38. Chang Y, He Y, Wang D, et al. ROS-regulated SUR1-TRPM4 drives persistent activation of NLRP3 inflammasome in microglia after whole-brain radiation. Acta Neuropathol Commun. 2025;13:16.
39. Sarkar S, Malovic E, Harishchandra DS, et al. Mitochondrial impairment in microglia amplifies NLRP3 inflammasome proinflammatory signaling in cell culture and animal models of Parkinson’s disease. NPJ Parkinson's Dis. 2017;3:30.
40. Simpson DSA, Oliver PL. ROS generation in microglia: understanding oxidative stress and inflammation in neurodegenerative disease. Antioxidants. 2020;9:743.
41. Gage MC, Thippeswamy T. Inhibitors of src family kinases, inducible nitric oxide synthase, and NADPH oxidase as potential CNS drug targets for neurological diseases. CNS Drugs. 2021;35:1-20.
42. Liu Z, Gan L, Zhang T, Ren Q, Sun C. Melatonin alleviates adipose inflammation through elevating α‐ketoglutarate and diverting adipose‐derived exosomes to macrophages in mice. J Pineal Res. 2017;64:e12455.
43. Lin Z, He H, Chen P, et al. Alpha-ketoglutarate protects against myocardial infarction via FTO-mediated anti-inflammatory macrophage activation. Basic Res Cardiol. 2025;120:889-912.
44. Zhou W, Hu G, He J, et al. SENP1-Sirt3 signaling promotes α-ketoglutarate production during M2 macrophage polarization. Cell Rep. 2022;39:110660.
45. Liu P, Wang H, Li X, et al. α-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat Immunol. 2017;18:985-94.
46. Zhu Y, Chen X, Lu Y, et al. Glutamine mitigates murine burn sepsis by supporting macrophage M2 polarization through repressing the SIRT5-mediated desuccinylation of pyruvate dehydrogenase. Burns Trauma. 2022;10:tkac041.
47. Agarwal S, Ghosh R, Verma G, Khadgawat R, Guchhait P. Alpha-ketoglutarate supplementation reduces inflammation and thrombosis in type 2 diabetes by suppressing leukocyte and platelet activation. Clin Exp Immunol. 2023;214:197-208.
48. Jia Y, Yin C, Ke W, et al. Alpha-ketoglutarate alleviates cadmium-induced inflammation by inhibiting the HIF1A-TNFAIP3 pathway in hepatocytes. Sci Total Environ. 2023;878:163069.
49. Zhao J, Kui L, Huang J, et al. Bifidobacterium animalis subsp. Lactis BX-BC08 modulates gut microbiota and secretes alpha-Ketoglutaric acid to alleviate MC903-induced atopic dermatitis. J Transl Med. 2025;23:768.
50. Joshi K, Liu S, Breslin SJP, Zhang J. Mechanisms that regulate the activities of TET proteins. Cell Mol Life Sci. 2022;79:363.
51. Cui X, Zheng Y, Lu Y, Issakidis-Bourguet E, Zhou D. Metabolic control of histone demethylase activity involved in plant response to high temperature. Plant Physiol. 2021;185:1813-28.
52. Ding R, Zhou Y, Zhang Q, et al. Regulation of α-ketoglutarate levels by Myc affects metabolism and demethylation in porcine early embryos. Front Cell Dev Biol. 2024;12:1507102.
53. Carey BW, Finley LWS, Cross JR, Allis CD, Thompson CB. Intracellular α-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature. 2014;518:413-6.
54. Szulwach KE, Li X, Li Y, et al. 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat Neurosci. 2011;14:1607-16.
55. Traube FR, Özdemir D, Sahin H, et al. Redirected nuclear glutamate dehydrogenase supplies Tet3 with α-ketoglutarate in neurons. Nat Commun. 2021;12:4100.
56. Ren X, Yan J, Zhao Q, et al. The Fe-S cluster assembly protein IscU2 increases α-ketoglutarate catabolism and DNA 5mC to promote tumor growth. Cell Discov. 2023;9:76.
57. Klemmensen MM, Borrowman SH, Pearce C, Pyles B, Chandra B. Mitochondrial dysfunction in neurodegenerative disorders. Neurotherapeutics. 2024;21:e00292.
58. Maurer I. A selective defect of cytochrome c oxidase is present in brain of Alzheimer disease patients. Neurobiol Aging. 2000;21:455-62.
59. Wang W, Zhao F, Lu Y, et al. Damaged mitochondria coincide with presynaptic vesicle loss and abnormalities in alzheimer’s disease brain. Acta Neuropathol Commun. 2023;11:54.
60. Henrich MT, Oertel WH, Surmeier DJ, Geibl FF. Mitochondrial dysfunction in Parkinson’s disease - a key disease hallmark with therapeutic potential. Mol Neurodegener. 2023;18:83.
61. Schapira AHV, Mann VM, Cooper JM, et al. Anatomic and disease specificity of NADH CoQ1 reductase (Complex I) deficiency in Parkinson's disease. J Neurochem. 2006;55:2142-5.
62. Bouter Y, Glasnek RM, Wenzel JM, Bouter C. 18F-FDG-PET and multimodal biomarker integration: a powerful tool for Alzheimer’s disease diagnosis. Nucl Med Mol Imaging. 2025;59:453-71.
63. Qin L, Crews FT. NADPH oxidase and reactive oxygen species contribute to alcohol-induced microglial activation and neurodegeneration. J Neuroinflammation. 2012;9:5.
64. Ganguly G, Chakrabarti S, Chatterjee U, Saso L. Proteinopathy, oxidative stress and mitochondrial dysfunction: cross talk in Alzheimer's disease and Parkinson's disease. Drug Des Devel Ther. 2017;11:797-810.
65. Thorne NJ, Tumbarello DA. The relationship of alpha-synuclein to mitochondrial dynamics and quality control. Front Mol Neurosci. 2022;15:947191.
66. Chen H, Chan DC. Mitochondrial dynamics-fusion, fission, movement, and mitophagy-in neurodegenerative diseases. Hum Mol Genet. 2009;18:R169-76.
67. Lacombe A, Scorrano L. The interplay between mitochondrial dynamics and autophagy: from a key homeostatic mechanism to a driver of pathology. Semin Cell Dev Biol. 2024;161-2:1-19.
68. Chen W, Zhao H, Li Y. Mitochondrial dynamics in health and disease: mechanisms and potential targets. Sig Transduct Target Ther. 2023;8:333.
69. Navakkode S, Kennedy BK. Alpha‐Ketoglutarate ameliorates synaptic plasticity deficits in APP/PS1 mice model of Alzheimer's disease. Aging Cell. 2025;24:e70235.
70. Yang X, Chen Y, Liu L, et al. Regulation of glycolysis-derived L-lactate production in astrocytes rescues the memory deficits and Aβ burden in early Alzheimer’s disease models. Pharmacol Res. 2024;208:107357.
71. Wang H, Fu J, Xu X, Yang Z, Zhang T. Rapamycin activates mitophagy and alleviates cognitive and synaptic plasticity deficits in a mouse model of Alzheimer’s disease. J Gerontol Series A. 2021;76:1707-13.
72. Su Y, Wang T, Wu N, et al. Alpha-ketoglutarate extends Drosophila lifespan by inhibiting mTOR and activating AMPK. Aging. 2019;11:4183-97.
73. Chin RM, Fu X, Pai MY, et al. The metabolite α-ketoglutarate extends lifespan by inhibiting ATP synthase and TOR. Nature. 2014;510:397-401.
74. Kostiuchenko O, Lushnikova I, Kowalczyk M, Skibo G. mTOR/α-ketoglutarate-mediated signaling pathways in the context of brain neurodegeneration and neuroprotection. BBA Adv. 2022;2:100066.
75. Johnson SC, Kayser E, Bornstein R, et al. Regional metabolic signatures in the Ndufs4(KO) mouse brain implicate defective glutamate/α-ketoglutarate metabolism in mitochondrial disease. Mol Genet Metab. 2020;130:118-32.
76. Neth BJ, Craft S. Insulin resistance and Alzheimer’s disease: bioenergetic linkages. Front Aging Neurosci. 2017;9:345.
77. Pawlosky RJ, Kashiwaya Y, King MT, Veech RL. A dietary ketone ester normalizes abnormal behavior in a mouse model of Alzheimer’s disease. Int J Mol Sci. 2020;21:1044.
78. Nicklas WJ, Vyas I, Heikkila RE. Inhibition of NADH-linked oxidation in brain mitochondria by 1-methyl-4-phenyl-pyridine, a metabolite of the neurotoxin, 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine. Life Sci. 1985;36:2503-8.
79. Li H, Zhang J, Shen Y, et al. Targeting mitochondrial complex I deficiency in MPP+/MPTP-induced Parkinson’s disease cell culture and mouse models by transducing yeast NDI1 gene. Biol Proced Online. 2024;26:9.
80. Satpute R, Lomash V, Kaushal M, Bhattacharya R. Neuroprotective effects of alpha-ketoglutarate and ethyl pyruvate against motor dysfunction and oxidative changes caused by repeated 1-methyl-4-phenyl-1,2,3,6 tetrahydropyridine exposure in mice. Hum Exp Toxicol. 2013;32:747-58.
81. Zhang W, Ding L, Zhang M, et al. Dietary intake of α-ketoglutarate ameliorates α-synuclein pathology in mouse models of Parkinson’s disease. Cell Mol Life Sci. 2023;80:155.
82. Hong S, Beja-Glasser VF, Nfonoyim BM, et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. 2016;352:712-6.
83. Pisalyaput K, Tenner AJ. Complement component C1q inhibits β‐amyloid‐ and serum amyloid P‐induced neurotoxicity via caspase‐ and calpain‐independent mechanisms. J Neurochem. 2007;104:696-707.
84. Webster SD, Yang AJ, Margol L, Garzon-Rodriguez W, Glabe CG, Tenner AJ. Complement component C1q modulates the phagocytosis of Aβ by microglia. Exp Neurol. 2000;161:127-38.
85. Woollard ML, Pearson RM, Dorf G, Griffith D, James IM. Controlled trial of ornithine alpha ketoglutarate (OAKG) in patients with stroke. Stroke. 1978;9:218-22.
86. Hua W, Zhang X, Tang H, et al. AKG attenuates cerebral ischemia-reperfusion injury through c‐Fos/IL‐10/Stat3 signaling pathway. Oxid Med Cell Longev. 2022;2022:6839385.
87. Tang Y, Han S, Asakawa T, et al. Effects of intracerebral hemorrhage on 5-hydroxymethylcytosine modification in mouse brains. Neuropsychiatr Dis Treat. 2016;2016:617.
88. Ding P, Zhu Q, Sheng B, et al. Alpha‐ketoglutarate alleviates neuronal apoptosis induced by central insulin resistance through inhibiting S6K1 Phosphorylation after subarachnoid hemorrhage. Oxid Med Cell Longev. 2022;2022:9148257.
89. Ari C, Poff AM, Held HE, et al. Metabolic therapy with deanna protocol supplementation delays disease progression and extends survival in amyotrophic lateral sclerosis (ALS) mouse model. PLoS ONE. 2014;9:e103526.
90. Allen SP, Al Sultan A, Kabucho Kibirige E, et al. A Y374X TDP43 truncation leads to an altered metabolic profile in amyotrophic lateral sclerosis fibroblasts driven by pyruvate and TCA cycle intermediate alterations. Front Aging Neurosci. 2023;15:1151848.
91. Ferreira IL, Cunha-Oliveira T, Nascimento MV, et al. Bioenergetic dysfunction in Huntington's disease human cybrids. Exp Neurol. 2011;231:127-34.
92. Niatsetskaya Z, Basso M, Speer RE, et al. HIF prolyl hydroxylase inhibitors prevent neuronal death induced by mitochondrial toxins: therapeutic implications for Huntington's disease and Alzheimer's disease. Antioxid Redox Signal. 2010;12:435-43.
93. Blackman G, Neri G, Al-Doori O, et al. Prevalence of neuroradiological abnormalities in first-episode psychosis: a systematic review and meta-analysis. JAMA Psychiatry. 2023;80:1047.
94. Opel N, Goltermann J, Hermesdorf M, Berger K, Baune BT, Dannlowski U. Cross-disorder analysis of brain structural abnormalities in six major psychiatric disorders: a secondary analysis of mega- and meta-analytical findings from the ENIGMA consortium. Biol Psychiatry. 2020;88:678-86.
95. Doucet GE, Janiri D, Howard R, O’brien M, Andrews-Hanna JR, Frangou S. Transdiagnostic and disease-specific abnormalities in the default-mode network hubs in psychiatric disorders: a meta-analysis of resting-state functional imaging studies. Eur Psychiatry. 2020;63:e57.
96. Ishida T, Nakamura Y, Tanaka SC, et al. Aberrant large-scale network interactions across psychiatric disorders revealed by large-sample multi-site resting-state functional magnetic resonance imaging datasets. Schizophr Bull. 2023;49:933-43.
97. Zhao Z, Li X, Xie Y, et al. Resolving the heterogeneity of dopamine subsystems dysfunction in schizophrenia: a PET meta-analysis. Schizophr. 2025;11:139.
98. Frankle WG, Himes M, Mason NS, Mathis CA, Narendran R. Prefrontal and striatal dopamine release are inversely correlated in schizophrenia. Biol Psychiatry. 2022;92:791-9.
99. Gluck MR, Thomas RG, Davis KL, Haroutunian V. Implications for altered glutamate and GABA metabolism in the dorsolateral prefrontal cortex of aged schizophrenic patients. Am J Psychiatry. 2002;159:1165-73.
100. Campbell IH, Campbell H. The metabolic overdrive hypothesis: hyperglycolysis and glutaminolysis in bipolar mania. Mol Psychiatry. 2024;29:1521-7.
101. Ressler KJ, Nemeroff CB. Role of serotonergic and noradrenergic systems in the pathophysiology of depression and anxiety disorders. Depress Anxiety. 2000;12:2-19.
102. Nunes PIG, Benjamin SR, Brito RDS, De Aguiar MR, Neves LB, De Bruin VMS. Mitochondria, oxidative stress, and psychiatric disorders: an integrative perspective on brain bioenergetics. Clin Bioenergetics. 2025;1:6.
103. Ni P, Ma Y, Chung S. Mitochondrial dysfunction in psychiatric disorders. Schizophr Res. 2024;273:62-77.
104. Townsend L, Pillinger T, Selvaggi P, Veronese M, Turkheimer F, Howes O. Brain glucose metabolism in schizophrenia: a systematic review and meta-analysis of 18FDG-PET studies in schizophrenia. Psychol Med. 2022;53:4880-97.
105. Rollins BL, Morgan L, Hjelm BE, et al. Mitochondrial complex I deficiency in schizophrenia and bipolar disorder and medication influence. Complex Psychiatry. 2017;3:157-69.
106. Bubber P, Hartounian V, Gibson G, Blass J. Abnormalities in the tricarboxylic acid (TCA) cycle in the brains of schizophrenia patients. Eur Neuropsychopharmacol. 2011;21:254-60.
107. Rangaraju V, Calloway N, Ryan TA. Activity-driven local ATP synthesis is required for synaptic function. Cell. 2014;156:825-35.
108. Davison J, O'gorman A, Brennan L, Cotter DR. A systematic review of metabolite biomarkers of schizophrenia. Schizophr Res. 2018;195:32-50.
109. Xuan J, Pan G, Qiu Y, et al. Metabolomic profiling to identify potential serum biomarkers for schizophrenia and risperidone action. J Proteome Res. 2011;10:5433-43.
110. Cai H, Li H, Yan X, et al. Metabolomic analysis of biochemical changes in the plasma and urine of first-episode neuroleptic-naïve schizophrenia patients after treatment with risperidone. J Proteome Res. 2012;11:4338-50.
111. Paredes RM, Quinones M, Marballi K, et al. Metabolomic profiling of schizophrenia patients at risk for metabolic syndrome. Int J Neuropsychopharm. 2014;17:1139-48.
112. Zhang W, Zhang M, Xu Z, et al. Human forebrain organoid-based multi-omics analyses of PCCB as a schizophrenia associated gene linked to GABAergic pathways. Nat Commun. 2023;14:5176.
113. Yoshimi N, Futamura T, Kakumoto K, et al. Blood metabolomics analysis identifies abnormalities in the citric acid cycle, urea cycle, and amino acid metabolism in bipolar disorder. BBA Clin. 2016;5:151-8.
114. Guo Q, Jia J, Sun XL, Yang H, Ren Y. Comparing the metabolic pathways of different clinical phases of bipolar disorder through metabolomics studies. Front Psychiatry. 2024;14:1319870.
115. Kaplan EN, Eren İ, Nazıroğlu M. Mood stabilizers (lamotrigine, lithium, and valproic acid) decrease bipolar disease model (ouabain)‐induced oxidative stress and apoptosis through the inhibition of the TRPM2 channel in neuronal cells. Bipolar Disord. 2025;27:483-500.
116. Eid F, El Ahmad P, Khoury R, et al. α-Ketoglutarate is a circulatory exercise factor that promotes learning and memory recall and has antidepressant properties. Biol Psychiatry Glob Open Sci. 2025;5:100477.
117. Tian J, Peng G, Gao X, et al. Dynamic analysis of the endogenous metabolites in depressed patients treated with TCM formula Xiaoyaosan using urinary 1H NMR-based metabolomics. J Ethnopharmacol. 2014;158:1-10.
118. Demianchuk O, Bayliak M, Vatashchuk M, et al. Alpha-ketoglutarate promotes anxiety, activates autophagy, and suppresses antioxidant enzymes in the cerebral cortex of female mice on cafeteria diet. Brain Res Bull. 2025;222:111255.
119. Ostrom QT, Price M, Neff C, et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2016-2020. Neuro Oncol. 2023;25:iv1-99.
120. Marin-Valencia I, Yang C, Mashimo T, et al. Analysis of tumor metabolism reveals mitochondrial glucose oxidation in genetically diverse human glioblastomas in the mouse brain in vivo. Cell Metab. 2012;15:827-37.
121. Deberardinis RJ, Mancuso A, Daikhin E, et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci USA. 2007;104:19345-50.
122. Yang C, Sudderth J, Dang T, Bachoo RG, Mcdonald JG, Deberardinis RJ. Glioblastoma cells require glutamate dehydrogenase to survive impairments of glucose metabolism or akt signaling. Cancer Res. 2009;69:7986-93.
123. Maus A, Peters GJ. Glutamate and α-ketoglutarate: key players in glioma metabolism. Amino Acids. 2016;49:21-32.
124. Ward PS, Patel J, Wise DR, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting α-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17:225-34.
125. Dang L, White DW, Gross S, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739-44.
126. Tönjes M, Barbus S, Park YJ, et al. BCAT1 promotes cell proliferation through amino acid catabolism in gliomas carrying wild-type IDH1. Nat Med. 2013;19:901-8.
127. Zhang B, Peng H, Zhou M, et al. Targeting BCAT1 combined with α-ketoglutarate triggers metabolic synthetic lethality in glioblastoma. Cancer Res. 2022;82:2388-402.
128. Lewis NA, Klein RH, Kelly C, Yee J, Knoepfler PS. Histone H3.3 K27M chromatin functions implicate a network of neurodevelopmental factors including ASCL1 and NEUROD1 in DIPG. Epigenetics Chromatin. 2022;15:18.
129. Lee K, Yun S, Park J, et al. Dimethyl alpha-ketoglutarate inhibits proliferation in diffuse intrinsic pontine glioma by reprogramming epigenetic and transcriptional networks. Biochem Biophys Res Commun. 2023;677:6-12.
130. Xu W, Yang H, Liu Y, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19:17-30.
131. Turcan S, Rohle D, Goenka A, et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. 2012;483:479-83.
132. Lang F, Kaur K, Fu H, et al. D-2-hydroxyglutarate impairs DNA repair through epigenetic reprogramming. Nat Commun. 2025;16:1431.
133. Wang X, Liu R, Qu X, et al. α-Ketoglutarate-activated NF-κB signaling promotes compensatory glucose uptake and brain tumor development. Mol Cell. 2019;76:148-62.e7.
134. Parker SJ, Encarnación-Rosado J, Hollinshead KER, et al. Spontaneous hydrolysis and spurious metabolic properties of α-ketoglutarate esters. Nat Commun. 2021;12:4905.
135. Baracco EE, Castoldi F, Durand S, et al. α-Ketoglutarate inhibits autophagy. Aging. 2019;11:3418-31.
136. Westi EW, Andersen JV, Aldana BI. Using stable isotope tracing to unravel the metabolic components of neurodegeneration: focus on neuron-glia metabolic interactions. Neurobiol Dis. 2023;182:106145.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].