


Membrane-bound or soluble, mesenchymal or myeloid: which RANKL drives osteoclastogenesis?
 0
0 Abstract
Receptor activator of nuclear factor-κB ligand (RANKL) is indispensable for osteoclast differentiation, activation, and bone resorption. While the relative contributions of membrane-bound RANKL (mRANKL) and soluble RANKL were historically debated, evidence from the past decade increasingly establishes mRANKL as the primary driver of osteoclastogenesis. However, a systematic literature synthesis integrating these diverse cellular sources and structural modalities remains lacking. It has also not been systematically defined which cellular sources of RANKL - arising from bone marrow mesenchymal lineage cells or hematopoietic lineage cells - play dominant roles in driving osteoclast activation. Clarifying these distinct forms and sources of RANKL will refine the conceptual framework of osteoclast regulation and provide a mechanistic basis for next-generation antiresorptive therapies that selectively target specific RANKL modalities to achieve more durable skeletal protection.
Keywords
INTRODUCTION
Osteoclast precursors originate from monocytes derived from hematopoietic stem cells. Their activation involves the phosphatidylinositol 3-kinase (PI3K)-protein kinase B (AKT)-mechanistic target of rapamycin (mTOR) and mitogen-activated protein kinase (MAPK)-extracellular signal-regulated kinase (ERK) signaling pathways. This activation occurs under stimulation by the macrophage colony-stimulating factor (M-CSF)-colony-stimulating factor 1 receptor (CSF1R) axis. These pathways drive the differentiation of monocytes into macrophages. Subsequently, macrophages further differentiate into osteoclasts in response to receptor activator of nuclear factor-κB ligand (RANKL) stimulation. During osteoclastogenesis, three major regulatory cascades operate: the M-CSF signaling pathway, the RANKL/receptor activator of nuclear factor-κB (RANK) /osteoprotegerin (OPG) axis, and the nuclear factor-κB (NF-κB) signaling pathway. Among them, the RANKL/RANK/OPG axis represents the most crucial and classical pathway governing osteoclast differentiation[1,2].
RANKL plays a central role in all stages of osteoclastogenesis. It regulates multiple key processes, including the fusion of osteoclast precursors into multinucleated cells, differentiation into mature osteoclasts, attachment to the bone surface, activation for bone resorption, and survival by preventing apoptosis. OPG, as the natural decoy receptor for RANKL, together with RANK and RANKL, forms the RANKL-RANK-OPG “tripartite regulatory system”, governing the rate of bone remodeling. An abnormal increase in the RANKL/OPG ratio within bone tissue is closely associated with pathological conditions such as osteoporosis, inflammatory bone destruction, and tumor-induced bone metastasis[3].
The RANKL/OPG axis has been successfully translated into clinical practice for the treatment of osteoporosis. Denosumab, a widely used anti-osteoporotic monoclonal antibody, mimics the endogenous function of OPG by neutralizing RANKL, thereby suppressing RANKL-dependent osteoclast differentiation and activation. However, in the absence of appropriate sequential therapy, its therapeutic effects dissipate rapidly after discontinuation. This often results in a pronounced “bone metabolism rebound”, characterized by sharply increased bone resorption, accelerated loss of bone mineral density, and, in severe cases, multiple vertebral compression fractures[3].
Therefore, elucidating the mechanisms through which the RANKL/OPG axis regulates osteoclast differentiation and function is essential for advancing its therapeutic translation. This review evaluates the spatiotemporal hierarchy of various cellular sources of tumor necrosis factor superfamily member 11 (Tnfsf11, mouse gene encoding RANKL) and clarifies the distinct, albeit secondary, role of soluble RANKL (sRANKL) in pathological bone resorption. We aim to provide a mechanistic framework for understanding why systemic Tnfsf11 blockade leads to metabolic rebound by examining the localized feedback loops between osteocytes and osteoclasts.
RANKL/OPG IN OSTEOCLAST ACTIVATION AND BONE RESORPTION
RANKL’s role in osteoclast activation
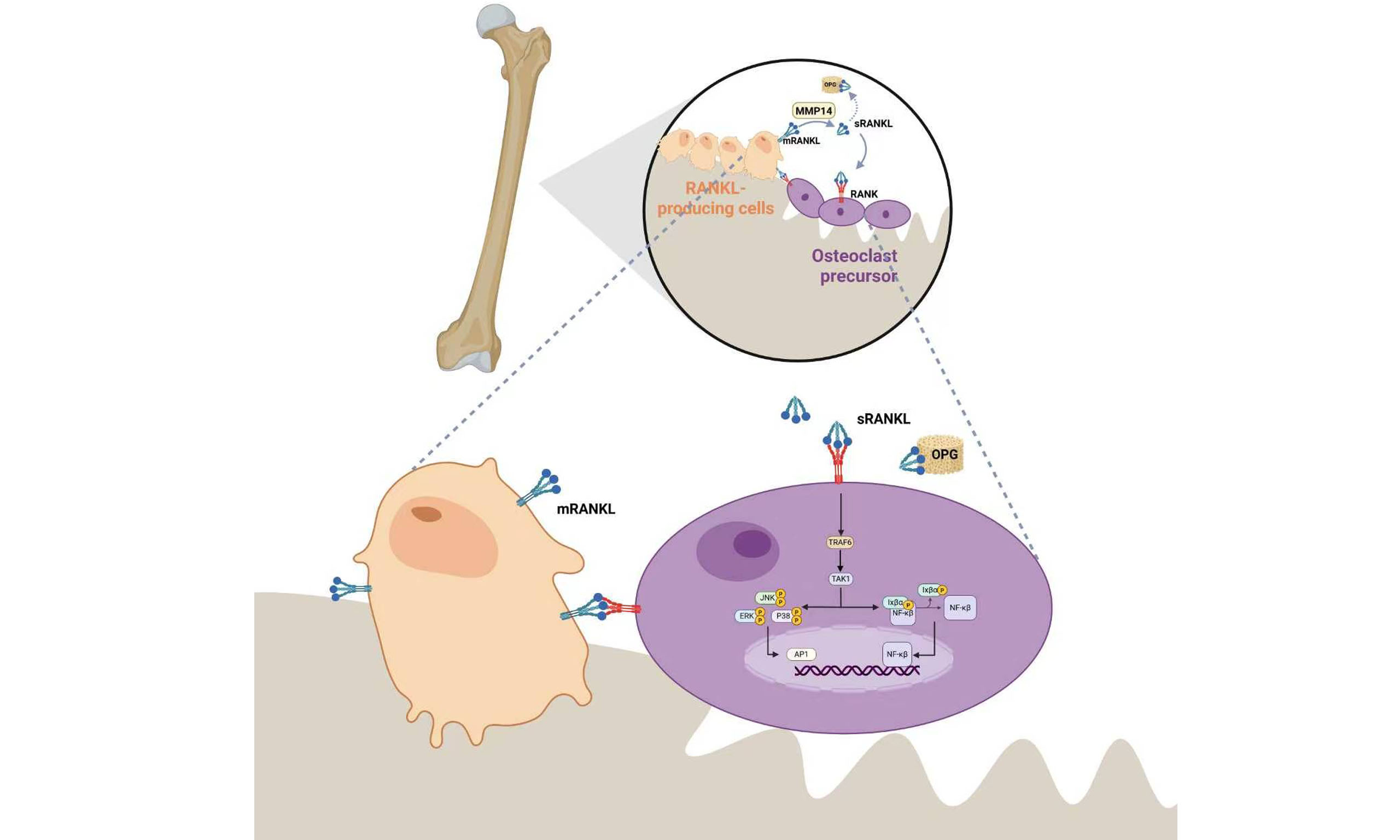
RANKL is a type II transmembrane protein encoded by a gene on human chromosome 13q14. It belongs to the tumor necrosis factor ligand family. The murine RANKL protein comprises 316 amino acids and shares approximately 85% sequence identity with its human ortholog, which contains 317 amino acids[4]. RANKL exists in both membrane-bound (40-45 kDa) and soluble (~31 kDa) forms, with the soluble form generated by proteolytic cleavage of the full-length protein near residues 140/145. RANKL is most abundantly expressed in bone tissue, but it is also detectable in several lymphoid organs, including lymph nodes, thymus, spleen, fetal liver, and Peyer’s patches[5].
RANKL plays a central role in bone homeostasis by regulating osteoclast differentiation and bone resorption. Its principal function is mediated by the binding of trimeric RANKL to homotrimeric RANK. This interaction induces receptor clustering and recruits tumor necrosis factor receptor associated factor 6 (TRAF6), thereby activating multiple downstream signaling cascades. These include the inhibitor of IκB kinase (IKK)/NF-κB, c-Jun N-terminal kinase (JNK)/activator protein-1 (AP-1), cellular myelocytomatosis oncogene (c-Myc), and calcineurin pathways, which collectively orchestrate osteoclast differentiation. In addition, the non-receptor tyrosine kinase Src and mitogen-activated protein kinase kinase 6-p38 mitogen-activated protein kinase (p38-MAPK)-melanocyte-inducing transcription factor pathways regulate osteoclast activation, whereas Src in conjunction with ERK promotes osteoclast survival[6-8]. Beyond the canonical kinase cascades, regulation of Ca2+ signaling in this pathway relies on co-stimulatory input mediated by immunoreceptor tyrosine-based activation motif (ITAM)[9,10].
OPG’s role in osteoclast activation
The OPG gene (Tnfrsf11b, tumor necrosis factor receptor superfamily member 11B) is located on human chromosome 8q23-24 and encodes a secreted, non-membrane-bound member of the tumor necrosis factor receptor (TNFR) superfamily. OPG is synthesized as a precursor of approximately 401 amino acids (aa) that includes a 21-aa signal peptide; removal of this signal peptide yields a mature polypeptide of ~380 aa. The N-terminal region of OPG contains four TNFR-like cysteine-rich domains (CRD1-CRD4). OPG is secreted predominantly as a disulfide-linked homodimer (~110 kDa), with a ~55 kDa monomer also detectable, and it is a glycoprotein that migrates at ~40 kDa after deglycosylation[11].
OPG acts as a soluble decoy receptor for RANKL. It directly binds the ligand, preventing its interaction with RANK, thereby suppressing osteoclastogenesis and bone resorption at their source[10]. Studies have found that the N-terminal CRDs mediate ligand recognition; truncation near Cysteine 185 abolishes activity, indicating that this segment is critical for RANKL neutralization[11]. Both mesenchymal and hematopoietic lineages have been established as important sources of OPG. This creates a local “sponge effect” for RANKL within the bone marrow, constraining its diffusion and reducing the likelihood of RANK engagement[6]. It is well established that multiple hormones and cytokines coordinately regulate expression of RANKL and OPG, thereby altering the RANKL/OPG ratio. An increase in this ratio augments osteoclast number and activity, whereas a decrease has the opposite effect, establishing the RANKL/OPG ratio as a functional rheostat of bone resorption[6,12].
mRANKL VS. sRANKL:WHICH FORM PLAYS THE DOMINANT ROLE IN OSTEOCLAST ACTIVATION
mRANKL’s role in osteoclast activation
To determine whether membrane-bound RANKL (mRANKL) or sRANKL predominantly regulates osteoclast differentiation, genetic approaches are essential, given their potential spatiotemporal and signaling differences. Xiong et al. used the clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 to delete amino acids isoleucine 133-serine 149 of the Tnfsf11 and substitute Arginine for the former tryptophan 150 residue, thereby generating a sheddase-resistant (SR) variant of the Tnfsf11[13]. This mouse strain was designated Tnfsf11SR. In Tnfsf11SR/SR mice, plasma sRANKL is undetectable, leaving only mRANKL. At 5 weeks of age, bone mass, tooth eruption, and the development of lymphoid organs and the mammary gland are essentially normal, indicating that mRANKL can execute most developmental functions of RANKL during the bone-modeling stage.
Further studies in 8-month-old Tnfsf11SR/SR mice showed that osteoclast numbers were reduced and trabecular bone mass increased, while cortical thickness remained unchanged. These results indicate that mRANKL is the principal driver of adult osteoclastogenesis/remodeling, whereas sRANKL primarily acts as an amplifier[13]. Accordingly, we speculate that the influence of sRANKL on skeletal development may increase with age. In addition, after undergoing ovariectomy (OVX), Tnfsf11SR/SR mice exhibit bone loss and increased bone remodeling comparable to wild-type controls, indicating that OVX-induced bone loss is independent of sRANKL and can be fully driven by mRANKL alone[13].
sRANKL’s role in osteoclast activation
sRANKL is generated primarily through proteolytic cleavage of the extracellular domain of mRANKL, and this ectodomain-shedding event is mediated by matrix metallopeptidase 14, a membrane-type matrix metalloproteinase[14]. Compared with mRANKL, sRANKL is dispensable during development but contributes to physiological trabecular bone remodeling in adulthood. Mizuno et al. generated two transgenic mouse lines overexpressing sRANKL and characterized their phenotypes[15]. Mice with global embryonic sRANKL overexpression died during late gestation, whereas mice in which sRANKL expression was restricted to the liver postnatally survived to adulthood. In the latter, trabecular bone began to decline by 6 weeks and was nearly absent by 3-4 months, while cortical bone remained largely unchanged[15]. Compared with OPG-deficient mice, bone loss in the sRANKL-overexpressing line progressed more slowly and was not accompanied by increased osteoblast numbers, suggesting that the impact of sRANKL on bone development is strongly age-dependent. Asano et al. generated a selectively sRANKL-deficient mouse model (Tnfsf11ΔS/ΔS) through targeted genetic deletion of its cleavage sites[16]. Their study demonstrated that sRANKL is physiologically dispensable for skeletal homeostasis and, importantly, does not drive the bone loss induced by estrogen deficiency[16].
Bone marrow mesenchymal lineage cell (BMSC)- VS. Hematopoietic stem cell (HSC)-DERIVED RANKL: WHICH SOURCE DRIVES OSTEOCLAST ACTIVATION
BMSC-derived RANKL
Prx1+ stem cells
Paired related homeobox-1 (Prx1)-positive cells mark early progenitors of the embryonic mesenchymal stem-cell lineage. They are localized primarily within the mesenchymal condensations of the limbs and parts of the craniofacial skeleton, but are largely absent from the axial skeleton and visceral organs[17].
Conditional deletion of RANKL in Prx1+ cells produces profound osteopetrosis in the appendicular long bones. At 5 weeks of age, trabecular bone volume fraction increases by approximately 4.5-fold, osteoclasts are nearly abolished, and hypertrophic cartilage persists at the growth plate, consistent with defective removal of calcified cartilage. In contrast, the spine is only minimally affected. These findings provide direct evidence that mesenchyme-derived RANKL is indispensable for osteoclastogenesis in developing long bones[17,18].
Because Prx1-cyclization recombination (Cre) is active in both chondrocyte and osteoblast lineages, the recombination encompasses both compartments. Further lineage-specific analyses are therefore required to delineate the respective contributions of these cell types [Figure 1].
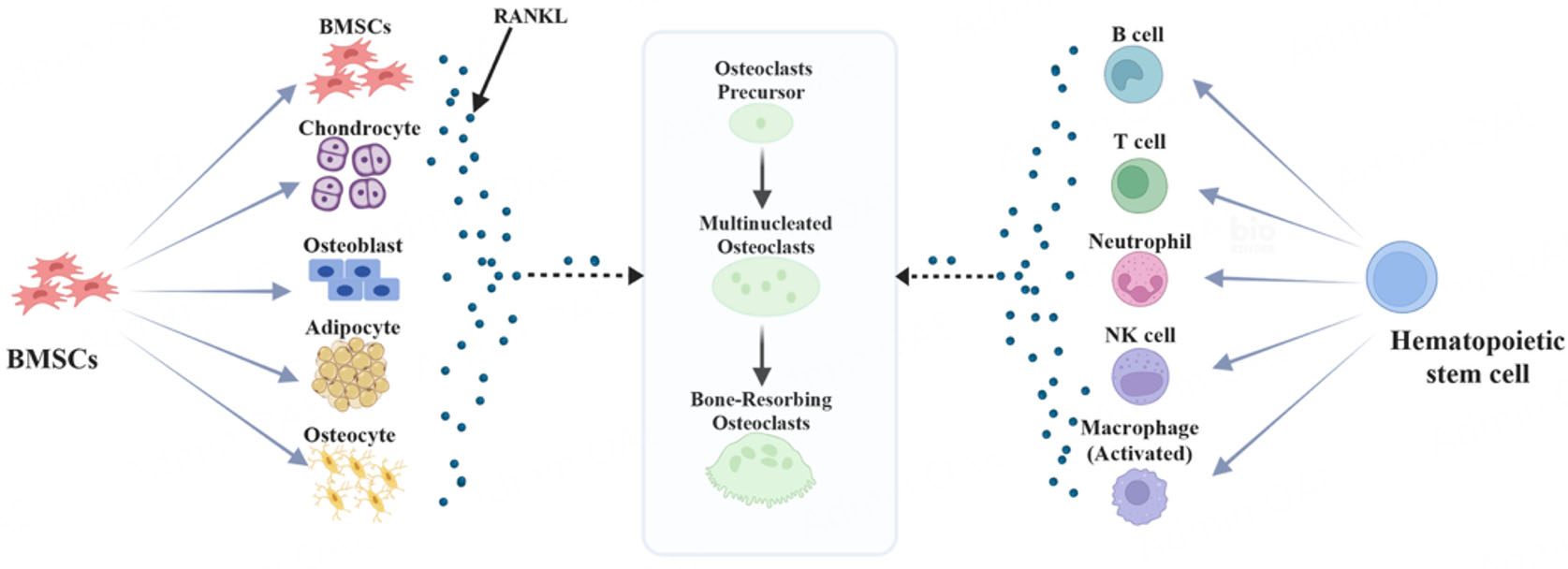
Figure 1. Schematic model illustrating how RANKL derived from BMSCs and HSCs regulates osteoclast precursor differentiation and activation. BMSCs: Bone marrow mesenchymal lineage cells; RANKL: receptor activator of nuclear factor-κB ligand. Created in BioRender. Sun, H. (2026) https://BioRender.com/6icl5f4.
Osteoblast
In the early stages of research, the hypothesis was proposed that osteoblasts and their precursors are the principal sources of RANKL in bone that facilitate osteoclast formation[19-21]. Osteoblast lineage and bone-marrow stromal cells constitute the most robust and consistent reservoir of RANKL; In combination with co-secreted M-CSF, they can drive the differentiation of monocytes into osteoclasts in vitro[22,23]. RANKL is predominantly membrane-bound and requires direct contact with osteoclast precursors; mechanical microdamage, parathyroid hormone, and 1,25-dihydroxyvitamin D3 [1,25(OH)2D3] have been shown to upregulate RANKL expression in osteoblast-lineage cells, thereby initiating osteoclastogenic programs.
Several in vivo studies have conditionally ablated matrix-producing osteoblasts to assess their role in bone resorption. Even when osteoblasts were completely depleted in mice and bone formation ceased, RANKL messenger RNA (mRNA) levels in bone remained unchanged over time, under both basal conditions and parathyroid hormone stimulation[24,25]. Osteoblast-lineage-specific ablation of RANKL - using osterix (Osx, also known as Sp7, encoded by the mouse gene Sp7) 1-Cre or osteocalcin (Ocn)-Cre - produces osteopetrosis[26-28]. At 5 weeks of age, mice exhibit an approximately four-fold increase in trabecular bone volume together with a marked reduction in osteoclasts within bone tissue. By contrast, the impact on adult bone remodeling is minimal: osteoclast numbers are not consistently reduced, osteopetrosis is not invariably induced, and osteoclast abundance during the remodeling phase remains unchanged. These findings may indicate that osteoclasts engaged in bone modeling are more sensitive to RANKL, or, alternatively, that osteoblast-derived RANKL is not an essential source during the remodeling phase[17] [Figure 1].
Chondrocyte
Chondrocytes - particularly those at the hypertrophic stage - express RANKL, and bone morphogenetic protein 2 stimulation robustly increases RANKL mRNA/protein, raising the RANKL/OPG ratio. In vitro, co-culture of chondrocytes with splenic osteoclast precursors from wild-type mice is sufficient to induce spontaneous osteoclast formation[29]. Moreover, 1,25(OH)2D3-VDR signaling induces RANKL in chondrocytes and promotes osteoclastogenesis both in vitro and in vivo. Conversely, loss of VDR in chondrocytes delays vascular invasion and osteoclast entry into the growth plate, indirectly indicating that the developmental “chondrocyte-to-osteoclast” sequence depends on a chondrocyte-derived RANKL pathway[30]. Consistent with this, selective deletion of Rankl in hypertrophic chondrocytes (collagen type X alpha 1 chain/Collagen X-Cre) leads to retention of mineralized cartilage, mirroring the growth-plate phenotype observed in Osx1/Ocn models[17,31]. Together, these findings demonstrate that RANKL from hypertrophic chondrocytes is a principal driver of mineralized cartilage clearance during endochondral development [Figure 1].
Osteocyte
Osteocytes modulate RANKL expression in neighboring cells in response to mechanical loading and injury cues. By sensing changes in load, they secrete RANKL to regulate osteoclastogenesis and cancellous bone remodeling, positioning osteocytes as a central source along the “mechanics-to-osteoclast” axis[17].
In vivo, osteocyte-specific deletion of RANKL using the dentin matrix protein 1 (Dmp1)-Cre driver shows no discernible difference in bone mass or osteoclast number at 5 weeks of age compared with littermate controls. Unlike Osx1-Cre or Ocn-Cre models[32-34], Dmp1-Cre-mediated deletion does not impair removal of calcified cartilage at the growth plate during the modeling phase. By approximately 6 months of age, however, Dmp1-Cre; Tnfsf11f/f mice exhibit a significant increase in bone mineral density. Specifically, the femoral trabecular bone volume fraction is roughly doubled, while the osteoclast surface is reduced by approximately 75%[17,18], effects comparable in magnitude to systemic RANKL blockade[35]. Moreover, in the hindlimb-unloading model, the knockout mice are markedly protected from bone loss, trabecular architectural deterioration, and cortical thinning; the unloading-induced rise in cortical RANKL observed in controls is absent, and the increase in osteoclast number is blunted[17]. Together, these findings identify osteocyte-derived RANKL as a pivotal driver of bone remodeling and of disuse-induced bone loss [Figure 1].
Bone marrow adipose lineage cells
Unlike peripheral adipose tissue, bone marrow adipose lineage cells (MALPs) arise from bone marrow mesenchymal lineage cells (BMSCs)[36,37]. Prior studies have shown that obesity and aging expand this adipose lineage within the marrow, thereby impairing both hematopoietic and skeletal regeneration[38]. Using adiponectin promoter-driven Cre (Adipoq-Cre) to selectively target MALPs, osteolineage-specific deletion of RANKL in Adipoq+ cells produced a modest increase in body weight relative to littermate controls. At 4, 8, and 16 weeks of age, these mice consistently showed increased trabecular bone in the femur and vertebrae, while cortical bone remained largely unaffected. Compared with Tnfsf11f/f controls, Adipoq-Cre; Tnfsf11f/f mice exhibited a marked reduction in osteoclast numbers within the marrow adjacent to trabeculae at
MALPs are cells that express adipogenic markers (e.g., Adipoq) yet lack lipid droplets and show minimal proliferative activity. Along the adipogenic trajectory, they occupy an intermediate position between mesenchymal progenitors and lipid-laden mature adipocytes[41]. To eliminate developmental confounders and define the role of MALP-derived RANKL in adult bone, Lu et al. generated inducible conditional knockout mice (Adipoq-CreER; Tnfsf11f/f, iCKO), enabling tamoxifen-triggered deletion of RANKL specifically in MALPs[42]. Inducible ablation of RANKL in adulthood rapidly increased trabecular bone mass in long bones and vertebrae and provided both prophylactic and therapeutic benefits in the OVX model of postmenopausal bone loss, while leaving cortical bone largely unchanged. Conversely, drill-hole injury repair was delayed, consistent with diminished coupling of bone formation to resorption[42] [Figure 1].
HSC-derived RANKL
B cell
Historically, the high expression of Tnfrsf11b in B lymphocytes at physiological steady state suggested they were a principal source of this decoy receptor. However, recent functional studies using conditional gene deletion have redefined this hierarchy[43]. Cawley et al. demonstrated that OPG derived from CD19+ B cells is physiologically dispensable for bone homeostasis, as its deletion failed to alter bone mass or cortical thickness[44]. Instead, mature osteoblasts emerged as the essential local source of OPG, directly suppressing RANKL activity to terminate the resorption phase of the bone remodeling cycle[44]. In contrast, B cells contribute little to the RANKL pool under homeostasis. Under pathological conditions such as estrogen deficiency, they transition into RANKL-expressing amplifiers, thereby exacerbating trabecular bone loss. Under estrogen deficiency and chronic low-grade inflammation, however, the B-cell compartment is remodeled: expression of RANKL and factors such as granulocyte colony-stimulating factor increases, the RANKL/OPG balance shifts downward, and B cells switch from “brakes” to drivers of osteoclastogenesis, thereby amplifying trabecular bone resorption[45,46].
In vivo, B cell-specific deletion of Tnfsf11 leaves baseline bone mass and turnover essentially unchanged in the absence of estrogen deficiency or inflammatory stimuli, indicating that B cell-derived RANKL is dispensable for steady-state remodeling. Following OVX, by contrast, B cell-specific Tnfsf11 deficiency confers robust protection against trabecular bone loss, with marked reductions in osteoclast number and osteoclast surface; this protective effect is largely confined to cancellous bone, with minimal impact on cortical bone[47].
The contribution of B cells to RANKL production is environment dependent; in estrogen-deficient or chronically inflamed marrow, they transition from OPG-dominant inhibitors to RANKL-expressing amplifiers of osteoclastogenesis. Consistent with this, B cell-specific loss of RANKL selectively mitigates OVX-induced trabecular bone loss while exerting little influence on physiologic homeostasis or cortical bone[46] [Figure 1].
T cell
In early postmenopausal women, the per-cell surface abundance of RANKL on bone-marrow T cells is markedly elevated and correlates positively with biochemical indices of bone resorption, indicating that under estrogen deficiency T cells can amplify osteoclastogenesis. In inflammatory arthritis models, activated T cells express RANKL and drive periarticular bone loss and erosion, underscoring T cells as one of the important sources of marrow RANKL activity[48].
In vivo, T cell-specific Tnfsf11 deletion leaves skeletal homeostasis essentially intact. Up to 7 months of age, whole-body and site-specific BMD are comparable to controls, and bone-marrow T/B-cell proportions are unaltered, implying that T cell-derived RANKL is not required for basal remodeling or modeling[47]. Moreover, T cell-specific Tnfsf11 deficiency does not mitigate OVX-induced bone loss. Declines in spinal, femoral, and whole-body BMD are similar to controls, and L4 trabecular bone volume/total volume (BV/TV) and trabecular thickness (Tb.Th), as well as femoral cortical thickness, show no improvement. This contrasts with B cell-specific Tnfsf11 deletion, which protects trabecular bone after OVX, pointing to a division of labor under estrogen deficiency. Consistent with earlier observations that mice lacking T cells are relatively resistant to OVX-induced bone loss, T cells clearly participate in postmenopausal bone catabolism; however, their principal contribution likely operates through cytokine networks (e.g., tumor necrosis factor (TNF)-α, interleukin (IL)-17) and indirect regulation of other cells (such as B cells and osteoblasts) rather than through T cell-intrinsic RANKL[49-52] [Figure 1].
Macrophage
In inflammatory bone-loss settings such as rheumatoid arthritis and periodontitis, macrophages can participate in - or themselves express - RANKL, thereby promoting focal osteoclastogenesis[53-56]. Macrophages broadly polarize into pro-inflammatory M1 and anti-inflammatory, pro-regenerative M2 phenotypes[57]. Multiple studies indicate that M1 macrophages inhibit osteogenic differentiation, whereas M2 macrophages more effectively support osteogenesis[58]. Co-culture of M1 macrophages with mesenchymal stem cells downregulates OPG, implying an indirect facilitation of osteoclast formation. Notably, M2 macrophages differentiate into mature osteoclasts in the presence of RANKL only under estrogen-deficient conditions. Collectively, the effects of macrophages on bone cells are polarization-dependent and modulated by estrogen [Figure 1].
NK cell
In rheumatoid arthritis (RA) synovial fluid and synovium, natural killer (NK) cells express RANKL together with M-CSF and are positioned in close proximity to CD14+ monocytes; in vitro, NK-monocyte co-cultures drive osteoclast differentiation in a RANKL- and M-CSF-dependent manner. In the murine collagen-induced arthritis model, depletion of NK cells nearly abolishes bone erosion[59], indicating that RANKL derived from NK cells can fuel inflammation-associated osteoclastic bone destruction. By contrast, during steady-state bone remodeling and in most systemic models, the dominant sources of RANKL remain osteocytes/osteoblastic lineage cells and MALPs [Figure 1].
Neutrophil
In RA, neutrophils isolated from synovial fluid and synovium have been shown to express RANKL at both the mRNA and protein levels and, in vitro, can directly drive monocytes to differentiate into osteoclasts[46,60]. Mechanistically, neutrophils secrete oncostatin M (OSM), which robustly induces RANKL expression in osteogenic-lineage cells[61]; moreover, neutrophil extracellular traps potentiate RANKL-induced osteoclastogenesis and thereby exacerbate bone erosion[62].
In vivo, authoritative genetic evidence defining the skeletal phenotype of a neutrophil-specific Tnfsf11 knockout is still lacking. Nevertheless, neutrophil depletion/inhibition studies - such as anti-lymphocyte antigen 6 complex locus G antibody treatment - in periodontitis and other inflammatory models consistently reduce RANKL+ cells, osteoclast numbers, and bone erosion[63], providing proof-of-concept and a strong rationale for future neutrophil-specific Tnfsf11 loss-of-function studies [Figure 1].
Spatiotemporal hierarchy and synergistic regulation of RANKL sources
The regulation of osteoclastogenesis relies on a sophisticated spatiotemporal hierarchy of RANKL sources that shifts across different skeletal life stages. During the bone modeling phase, RANKL derived from hypertrophic chondrocytes and early osteoblast-lineage cells serves as the primary driver[17,29]. Specifically, chondrocyte-derived RANKL is essential for the clearance of mineralized cartilage during endochondral ossification, while osteoblasts provide the requisite signals for initial bone modeling in the appendicular skeleton.
As the skeleton transitions into the adult remodeling phase, the dominance of RANKL production shifts toward matrix-embedded osteocytes and MALPs. Osteocytes, acting as mechanosensors, provide the pivotal RANKL signals required for steady-state remodeling and disuse-induced bone loss. Concurrently[18], MALPs emerge as a major regulator of trabecular bone turnover, particularly in the context of aging and metabolic changes[39,40].
This mesenchymal-derived RANKL framework does not operate in isolation but synergizes with local immune cells to fine-tune bone resorption[43]. Under physiological steady state, hematopoietic cells such as B cells primarily exert an anti-resorptive influence by maintaining a high OPG/RANKL ratio. However, in pathological contexts such as estrogen deficiency or chronic inflammation, the bone marrow microenvironment facilitates a “role reversal”: B cells and activated T cells transition into potent RANKL-expressing amplifiers. The synergy between mesenchymal “commanders” (osteocytes/MALPs) and hematopoietic “amplifiers” (B/T cells) creates a hyper-resorptive milieu. This provides a mechanistic explanation for the rapid bone loss observed in postmenopausal osteoporosis and inflammatory bone diseases[46,47] [Figure 1].
REGULATORY ROLE OF EXTRA-SKELETAL-DERIVED SRANKL IN OSTEOCLAST FUNCTION
As a secreted protein, sRANKL is also produced by various extra-skeletal organs and tissues, enters the circulation, and subsequently reaches bone, where it contributes to the regulation of osteoclast-mediated bone resorption.
Activated human T cells can produce a secreted splice variant of tumor necrosis factor ligand superfamily member 11 (TNFSF11, human gene encoding RANKL) that yields sRANKL, which by itself is sufficient to support osteoclastogenesis. In parallel, T cells can release sRANKL via proteolytic ectodomain shedding of membrane RANKL[64]. In periodontitis and related inflammatory settings, activated B/lymphoid cells generate sRANKL through TNF-α converting enzyme/A disintegrin and metalloproteinase domain 17-mediated shedding, thereby amplifying osteoclast formation[65]. Rheumatoid arthritis synovial fibroblasts overexpress RANKL and secrete osteoclastogenic mediators; their conditioned media can drive peripheral blood monocytes to differentiate into osteoclasts, positioning the synovial lesion as a major source of sRANKL[66].
Human microvascular endothelial cells upregulate RANKL in response to inflammatory cytokines such as IL-1 and TNF-α, thereby promoting osteoclast formation[67]. Concurrently, they increase the expression of intercellular adhesion molecule 1, vascular cell adhesion molecule 1, and CD44, which markedly enhances the adhesion and transendothelial migration of circulating CD14+ osteoclast precursors, indirectly amplifying osteoclastogenesis and bone resorption at sites of inflammation[68].
Multiple hepatic cell types - including hepatocytes, Kupffer cells, liver sinusoidal endothelial cells, and hepatic stellate cells - express RANKL and its decoy receptor OPG[69]. Under pathological conditions such as chronic liver disease, trauma, or systemic inflammation, hepatic RANKL expression is typically induced in parallel with inflammatory activation. Histologically, increased densities of RANKL-positive cells are observed within portal tracts, along fibrous septa, and around bile ducts. Upregulated RANKL can be shed or otherwise released into the circulation, thereby contributing to the systemic pool of sRANKL and amplifying osteoclastogenic signaling[69-71]. Consistent with this, several studies have reported elevated serum sRANKL in non-cirrhotic stages of chronic liver disease[70]. Nevertheless, because bone remodeling is governed predominantly by local paracrine interactions, circulating RANKL/OPG concentrations only partially capture the effective signaling milieu within bone.
Tumor-associated cells also constitute important sources of sRANKL that promote local-and in some cases systemic-bone resorption. In multiple myeloma, malignant plasma cells themselves produce sRANKL, correlating with elevated circulating sRANKL and generalized bone loss[72,73]. In addition, myeloma cells stimulate bone marrow stromal/osteoblastic cells to increase RANKL, creating a malignant bone-tumor feed-forward loop. Breast and prostate cancer cells increase the RANKL/OPG ratio in the bone microenvironment directly or indirectly (e.g., via parathyroid hormone-related protein, IL-6), thereby amplifying osteoclastogenesis, driving osteolysis, and facilitating skeletal metastasis[16,74-76].
RANKL REVERSE SIGNALING: FROM OSTEOCLAST TO OSTEOBLAST AND BACK TO OSTEOCLAST
Beyond the functions described above, RANKL acts as a bidirectional signaling molecule. The canonical “forward” pathway is the RANKL-RANK interaction that promotes osteoclastogenesis. By contrast, “reverse” signaling is initiated within RANKL-expressing cells when multiple RANKL trimers are cross-linked into higher-order clusters. The cytoplasmic tail of RANKL contains proline-rich motifs that engage Src family kinases, thereby activating downstream pathways. In bone, vesicular RANK derived from osteoclasts can serve as a ligand to engage RANKL on osteoblasts and trigger RANKL reverse signaling. This signaling drives runt-related transcription factor 2 nuclear translocation and early osteoblast differentiation, thereby supporting the coupling between bone resorption and formation[77]. RANKL is also subject to intrinsic negative feedback. In osteoclast precursors, RANKL induces expression of the deubiquitinase ubiquitin C-terminal hydrolase L1 (UCHL1), which directly interacts with CD13 to deubiquitinate and stabilize the CD13 protein, thereby promoting the release of soluble CD13 (sCD13). Acting in an autocrine manner, sCD13 attenuates RANKL-driven MAPK signaling (JNK/ERK/p38) with minimal impact on NF-κB, ultimately suppressing osteoclast differentiation. This mechanism provides a rationale for how, in the early stages of osteoarthritis, restricting osteoclastogenesis within the subchondral bone compartment can help protect the joint[78].
DISCUSSION
The relative contributions of mRANKL and sRANKL to osteoclastogenesis have been historically debated. However, recent pivotal genetic studies have increasingly established mRANKL as the primary driver of both skeletal development and adult bone remodeling[13]. Our synthesis confirms that mice selectively lacking sRANKL (Tnfsf11ΔS/ΔS) exhibit normal bone density and skeletal microstructure, demonstrating that sRANKL is physiologically dispensable for basal homeostasis[16]. In contrast, mRANKL serves as the indispensable ligand for physiological bone turnover and the accelerated resorption observed in estrogen-deficiency models. While sRANKL appears redundant for normal skeletal health, its role is markedly prominent in pathological settings. It acts as a chemotactic agent that facilitates the migration of RANK-expressing tumor cells to bone, thereby promoting skeletal metastasis without directly influencing osteoclast activation[16].
The clinical translation of the RANKL-RANK-OPG axis, exemplified by Denosumab, has encountered significant challenges, most notably the “rebound bone turnover” following treatment cessation[3]. Mechanistically, the withdrawal of RANKL inhibition leads to a localized surge in Tnfsf11 levels, triggering the rapid fusion of accumulated osteoclast precursors - termed osteomorphs - into active, bone-resorbing cells[2]. This metabolic burst, compounded by an insufficient compensatory OPG response, significantly elevates the risk of multiple vertebral compression fractures. To mitigate these risks, next-generation antiresorptive strategies must move toward precision targeting[3].
FUTURE PERSPECTIVES
Despite substantial progress, several conceptual and translational gaps remain. A key priority is to delineate the spatiotemporal dynamics of RANKL production across mesenchymal and hematopoietic compartments, particularly in the contexts of aging, metabolic dysregulation, malignancy, and chronic inflammation. Advances in single-cell multi-omics and spatial transcriptomics are expected to uncover previously unrecognized microanatomical niches that fine-tune RANKL accessibility during skeletal remodeling.
Another promising direction is to define the functional significance of RANKL reverse signaling within osteoblast-lineage cells, which may represent an underexplored mechanism governing the coupling of bone resorption and formation. Denosumab, a potent full-spectrum RANKL-neutralizing antibody, is frequently associated with a pronounced “rebound bone turnover” upon discontinuation, resulting in a rapid loss of bone mineral density[3]. Recent mechanistic insights reveal that this phenomenon is driven by a localized surge in RANKL levels following treatment withdrawal, which triggers the swift fusion of long-term suppressed osteoclast precursors (osteomorphs) into active bone-resorbing cells[2]. Furthermore, emerging evidence underscores the regulatory role of osteocyte-derived OPG; an insufficient OPG response fails to antagonize the excessive RANKL signaling, thereby exacerbating this metabolic dysregulation. Clinically, these abrupt fluctuations in bone turnover significantly elevate the risk of multiple vertebral compression fractures. To mitigate these skeletal events, current management strategies advocate for the immediate sequential administration of anti-resorptive agents, such as bisphosphonates, to dampen the transient burst in osteoclast activity[3]. Additionally, the identification of the RANKL-UCHL1-sCD13 negative feedback loop provides a promising molecular target. Targeting this loop could help stabilize subchondral bone and prevent systemic bone loss[78]. Looking forward, the development of precision therapies is essential; as proposed by Asano et al., selective inhibitors targeting sRANKL represent a superior anti-metastatic strategy[16]. By inhibiting sRANKL-mediated tumor cell migration while preserving the physiologically indispensable mRANKL, such agents are hypothesized to carry fewer systemic adverse effects and minimize the risks associated with total RANKL blockade[16]. From a therapeutic standpoint, next-generation antiresorptive strategies should prioritize precision targeting - such as selectively blocking RANKL derived from specific cellular sources (e.g., MALPs or osteocytes) or preferentially inhibiting the soluble form - to minimize adverse effects and prevent systemic rebound in bone turnover.
CONCLUSION
A comprehensive body of genetic, cellular, and lineage-tracing evidence demonstrates that the RANKL/RANK/OPG axis serves as the central regulatory system governing osteoclast differentiation, activation, and skeletal turnover[1,2]. This review integrates decades of research delineating the distinct contributions of mRANKL and sRANKL, as well as the diverse cellular sources of RANKL within the bone marrow and across extra-skeletal organs and tissues. Collectively, current evidence establishes that mRANKL is indispensable for developmental osteoclastogenesis and remains the predominant driver of physiological bone remodeling in adulthood, whereas sRANKL functions mainly as an age-dependent amplifier of trabecular bone resorption[13-15].
Advances in conditional knockout models have further refined the hierarchy of RANKL sources, identifying osteocytes and MALPs as key regulators of adult skeletal remodeling[17,39,40]. In addition, extra-skeletal tissues-including lymphoid organs, the liver, vascular endothelium, and various tumors-contribute to circulating sRANKL and may thereby influence systemic osteoclastogenic activity. Together, these findings redefine current concepts of RANKL-mediated osteoclast regulation and provide an updated mechanistic framework for understanding both physiological and pathological bone resorption.
DECLARATION
Acknowledgments
The graphical abstract was created with BioRender.com (Created in BioRender. Feng, Y. (2026) https://BioRender.com/0ptj2vp).
Authors’ contributions
Responsible for the conceptualization, structural design, and initial drafting of the manuscript: Zhang T, Feng Y, Wang W (Wei Wang)
Conducted the literature search, performed data extraction from the included studies, and assisted in the synthesis of recent genetic evidence: Wang W (Wenzhao Wang)
Contributed to the critical revision of the manuscript for important intellectual content, particularly regarding clinical translation and future perspectives: Shan L
Responsible for the final approval of the version to be published and supervised the entire review process: Yang X, Sun H, Feng S
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
During the preparation of this manuscript, the AI tool ChatGPT (version 5.0, released 2025-08-07) was used solely for language editing. The tool did not influence the study design, data collection, analysis, interpretation, or the scientific content of the work. All authors take full responsibility for the accuracy, integrity, and final content of the manuscript.
Financial support and sponsorship
This work was supported by the National Natural Science Foundation of China (Grant No. 82401025), the Zhejiang Provincial Natural Science Foundation of China (Grant No. LQN26H060004), and the China Postdoctoral Science Foundation (Grant No. 2025M772006).
Conflicts of interest
Sun H and Wang W (Wenzhao Wang) are Young Editorial Board Members of Journal of Translational Genetics and Genomics. They were not involved in any steps of the editorial process for this manuscript, including reviewers’ selection, manuscript handling, or decision-making, while the other authors have declared that they have no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Wu Q, Liang H, Wang C, et al. Tetrahydroberberine prevents ovariectomy-induced bone loss by inhibiting RANKL-induced osteoclastogenesis and promoting osteoclast apoptosis. J Agric Food Chem. 2024;72:20383-95.
2. Kim AS, Taylor VE, Castro-Martinez A, et al. Temporal patterns of osteoclast formation and activity following withdrawal of RANKL inhibition. J Bone Miner Res. 2024;39:484-97.
3. Kumar S, Wang M, Kim AS, Center JR, McDonald MM, Girgis CM. Denosumab discontinuation in the clinic: implications of rebound bone turnover and emerging strategies to prevent bone loss and fractures. J Bone Miner Res. 2025;40:1017-34.
4. Anderson DM, Maraskovsky E, Billingsley WL, et al. A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature. 1997;390:175-9.
6. Boyce BF, Xing L. Functions of RANKL/RANK/OPG in bone modeling and remodeling. Arch Biochem Biophys. 2008;473:139-46.
7. Lomaga MA, Yeh WC, Sarosi I, et al. TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes Dev. 1999;13:1015-24.
8. Naito A, Azuma S, Tanaka S, et al. Severe osteopetrosis, defective interleukin-1 signalling and lymph node organogenesis in TRAF6-deficient mice. Genes Cells. 1999;4:353-62.
9. Koga T, Inui M, Inoue K, et al. Costimulatory signals mediated by the ITAM motif cooperate with RANKL for bone homeostasis. Nature. 2004;428:758-63.
10. Udagawa N, Koide M, Nakamura M, et al. Osteoclast differentiation by RANKL and OPG signaling pathways. J Bone Miner Metab. 2021;39:19-26.
11. Simonet WS, Lacey DL, Dunstan CR, et al. Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell. 1997;89:309-19.
12. Rao S, Cronin SJF, Sigl V, Penninger JM. RANKL and RANK: from mammalian physiology to cancer treatment. Trends Cell Biol. 2018;28:213-23.
13. Xiong J, Cawley K, Piemontese M, et al. Soluble RANKL contributes to osteoclast formation in adult mice but not ovariectomy-induced bone loss. Nat Commun. 2018;9:2909.
14. Hikita A, Yana I, Wakeyama H, et al. Negative regulation of osteoclastogenesis by ectodomain shedding of receptor activator of NF-kappaB ligand. J Biol Chem. 2006;281:36846-55.
15. Mizuno A, Kanno T, Hoshi M, et al. Transgenic mice overexpressing soluble osteoclast differentiation factor (sODF) exhibit severe osteoporosis. J Bone Miner Metab. 2002;20:337-44.
16. Asano T, Okamoto K, Nakai Y, et al. Soluble RANKL is physiologically dispensable but accelerates tumour metastasis to bone. Nat Metab. 2019;1:868-75.
17. Xiong J, Onal M, Jilka RL, Weinstein RS, Manolagas SC, O'Brien CA. Matrix-embedded cells control osteoclast formation. Nat Med. 2011;17:1235-41.
18. Xiong J, O'Brien CA. Osteocyte RANKL: new insights into the control of bone remodeling. J Bone Miner Res. 2012;27:499-505.
19. Kearns AE, Khosla S, Kostenuik PJ. Receptor activator of nuclear factor kappaB ligand and osteoprotegerin regulation of bone remodeling in health and disease. Endocr Rev. 2008;29:155-92.
20. Sims NA, Gooi JH. Bone remodeling: multiple cellular interactions required for coupling of bone formation and resorption. Semin Cell Dev Biol. 2008;19:444-51.
22. Suda T, Takahashi N, Udagawa N, Jimi E, Gillespie MT, Martin TJ. Modulation of osteoclast differentiation and function by the new members of the tumor necrosis factor receptor and ligand families. Endocr Rev. 1999;20:345-57.
23. Bord S, Ireland DC, Beavan SR, Compston JE. The effects of estrogen on osteoprotegerin, RANKL, and estrogen receptor expression in human osteoblasts. Bone. 2003;32:136-41.
24. Galli C, Fu Q, Wang W, et al. Commitment to the osteoblast lineage is not required for RANKL gene expression. J Biol Chem. 2009;284:12654-62.
25. Corral DA, Amling M, Priemel M, et al. Dissociation between bone resorption and bone formation in osteopenic transgenic mice. Proc Natl Acad Sci USA. 1998;95:13835-40.
26. Zhang M, Xuan S, Bouxsein ML, et al. Osteoblast-specific knockout of the insulin-like growth factor (IGF) receptor gene reveals an essential role of IGF signaling in bone matrix mineralization. J Biol Chem. 2002;277:44005-12.
27. Kobayashi T, Lu J, Cobb BS, et al. Dicer-dependent pathways regulate chondrocyte proliferation and differentiation. Proc Natl Acad Sci USA. 2008;105:1949-54.
28. Rodda SJ, McMahon AP. Distinct roles for Hedgehog and canonical Wnt signaling in specification, differentiation and maintenance of osteoblast progenitors. Development. 2006;133:3231-44.
29. Usui M, Xing L, Drissi H, et al. Murine and chicken chondrocytes regulate osteoclastogenesis by producing RANKL in response to BMP2. J Bone Miner Res. 2008;23:314-25.
30. Masuyama R, Stockmans I, Torrekens S, et al. Vitamin D receptor in chondrocytes promotes osteoclastogenesis and regulates FGF23 production in osteoblasts. J Clin Invest. 2006;116:3150-9.
31. Gebhard S, Hattori T, Bauer E, et al. Specific expression of Cre recombinase in hypertrophic cartilage under the control of a BAC-Col10a1 promoter. Matrix Biol. 2008;27:693-9.
32. Kalajzic I, Braut A, Guo D, et al. Dentin matrix protein 1 expression during osteoblastic differentiation, generation of an osteocyte GFP-transgene. Bone. 2004;35:74-82.
33. Yang W, Lu Y, Kalajzic I, et al. Dentin matrix protein 1 gene cis-regulation: use in osteocytes to characterize local responses to mechanical loading in vitro and in vivo. J Biol Chem. 2005;280:20680-90.
34. Lu Y, Xie Y, Zhang S, Dusevich V, Bonewald LF, Feng JQ. DMP1-targeted Cre expression in odontoblasts and osteocytes. J Dent Res. 2007;86:320-5.
35. Kong YY, Yoshida H, Sarosi I, et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature. 1999;397:315-23.
36. Zhou BO, Yu H, Yue R, et al. Bone marrow adipocytes promote the regeneration of stem cells and haematopoiesis by secreting SCF. Nat Cell Biol. 2017;19:891-903.
37. Zhou BO, Yue R, Murphy MM, Peyer JG, Morrison SJ. Leptin-receptor-expressing mesenchymal stromal cells represent the main source of bone formed by adult bone marrow. Cell Stem Cell. 2014;15:154-68.
38. Ambrosi TH, Scialdone A, Graja A, et al. Adipocyte accumulation in the bone marrow during obesity and aging impairs stem cell-based hematopoietic and bone regeneration. Cell Stem Cell. 2017;20:771-84.e6.
39. Hu Y, Li X, Zhi X, et al. RANKL from bone marrow adipose lineage cells promotes osteoclast formation and bone loss. EMBO Rep. 2021;22:e52481.
40. Yu W, Zhong L, Yao L, et al. Bone marrow adipogenic lineage precursors promote osteoclastogenesis in bone remodeling and pathologic bone loss. J Clin Invest. 2021;131:e140214.
41. Zhong L, Yao L, Tower RJ, et al. Single cell transcriptomics identifies a unique adipose lineage cell population that regulates bone marrow environment. Elife. 2020;9:e54695.
42. Lu J, He Q, Wang H, et al. Bone marrow adipogenic lineage precursors are the major regulator of bone resorption in adult mice. Bone Res. 2025;13:39.
43. Walsh MC, Choi Y. Biology of the RANKL-RANK-OPG system in immunity, bone, and beyond. Front Immunol. 2014;5:511.
44. Cawley KM, Bustamante-Gomez NC, Guha AG, et al. Local production of osteoprotegerin by osteoblasts suppresses bone resorption. Cell Rep. 2020;32:108052.
45. Weitzmann MN. The role of inflammatory cytokines, the RANKL/OPG axis, and the immunoskeletal interface in physiological bone turnover and osteoporosis. Scientifica. 2013;2013:125705.
46. Fischer V, Haffner-Luntzer M. Interaction between bone and immune cells: Implications for postmenopausal osteoporosis. Semin Cell Dev Biol. 2022;123:14-21.
47. Onal M, Xiong J, Chen X, et al. Receptor activator of nuclear factor κB ligand (RANKL) protein expression by B lymphocytes contributes to ovariectomy-induced bone loss. J Biol Chem. 2012;287:29851-60.
48. Eghbali-fatourechi G, Khosla S, Sanyal A, Boyle WJ, Lacey DL, Riggs BL. Role of RANK ligand in mediating increased bone resorption in early postmenopausal women. J Clin Investig. 2003;111:1221-30.
49. Xu J, Yu L, Liu F, Wan L, Deng Z. The effect of cytokines on osteoblasts and osteoclasts in bone remodeling in osteoporosis: a review. Front Immunol. 2023;14:1222129.
50. Wu D, Cline-Smith A, Goering D, Choudhary A, Veis D, Aurora R. Estrogen loss activates memory T-cells to compromise bone integrity through distinct cortical compartments in mice. J Bone Miner Res. 2025;40:1087-99.
51. Huang F, Wong P, Li J, et al. Osteoimmunology: the correlation between osteoclasts and the Th17/Treg balance in osteoporosis. J Cell Mol Med. 2022;26:3591-7.
52. Bhadricha H, Patel V, Singh AK, et al. Increased frequency of Th17 cells and IL-17 levels are associated with low bone mineral density in postmenopausal women. Sci Rep. 2021;11:16155.
53. Kittaka M, Yoshimoto T, Levitan ME, et al. Osteocyte RANKL drives bone resorption in mouse ligature-induced periodontitis. J Bone Miner Res. 2023;38:1521-40.
54. Crotti TN, Smith MD, Weedon H, et al. Receptor activator NF-kappaB ligand (RANKL) expression in synovial tissue from patients with rheumatoid arthritis, spondyloarthropathy, osteoarthritis, and from normal patients: semiquantitative and quantitative analysis. Ann Rheum Dis. 2002;61:1047-54.
55. Ono T, Hayashi M, Sasaki F, Nakashima T. RANKL biology: bone metabolism, the immune system, and beyond. Inflamm Regen. 2020;40:2.
57. Clark D, Brazina S, Yang F, et al. Age-related changes to macrophages are detrimental to fracture healing in mice. Aging Cell. 2020;19:e13112.
58. Zhang Y, Böse T, Unger RE, Jansen JA, Kirkpatrick CJ, van den Beucken JJJP. Macrophage type modulates osteogenic differentiation of adipose tissue MSCs. Cell Tissue Res. 2017;369:273-86.
59. Söderström K, Stein E, Colmenero P, et al. Natural killer cells trigger osteoclastogenesis and bone destruction in arthritis. Proc Natl Acad Sci USA. 2010;107:13028-33.
60. Poubelle PE, Chakravarti A, Fernandes MJ, Doiron K, Marceau AA. Differential expression of RANK, RANK-L, and osteoprotegerin by synovial fluid neutrophils from patients with rheumatoid arthritis and by healthy human blood neutrophils. Arthritis Res Ther. 2007;9:R25.
61. Ando Y, Tsukasaki M, Huynh NC, et al. The neutrophil-osteogenic cell axis promotes bone destruction in periodontitis. Int J Oral Sci. 2024;16:18.
62. Schneider AH, Taira TM, Públio GA, et al. Neutrophil extracellular traps mediate bone erosion in rheumatoid arthritis by enhancing RANKL-induced osteoclastogenesis. Br J Pharmacol. 2024;181:429-46.
63. Kim AR, Kim JH, Choi YH, et al. The presence of neutrophils causes RANKL expression in periodontal tissue, giving rise to osteoclast formation. J Periodontal Res. 2020;55:868-76.
64. Walsh NC, Alexander KA, Manning CA, et al. Activated human T cells express alternative mRNA transcripts encoding a secreted form of RANKL. Genes Immun. 2013;14:336-45.
65. Kanzaki H, Makihira S, Suzuki M, et al. Soluble RANKL cleaved from activated lymphocytes by TNF-α-converting enzyme contributes to osteoclastogenesis in periodontitis. J Immunol. 2016;197:3871-83.
66. Dickerson TJ, Suzuki E, Stanecki C, Shin HS, Qui H, Adamopoulos IE. Rheumatoid and pyrophosphate arthritis synovial fibroblasts induce osteoclastogenesis independently of RANKL, TNF and IL-6. J Autoimmun. 2012;39:369-76.
67. Collin-Osdoby P, Rothe L, Anderson F, Nelson M, Maloney W, Osdoby P. Receptor activator of NF-kappa B and osteoprotegerin expression by human microvascular endothelial cells, regulation by inflammatory cytokines, and role in human osteoclastogenesis. J Biol Chem. 2001;276:20659-72.
68. Kindle L, Rothe L, Kriss M, Osdoby P, Collin-Osdoby P. Human microvascular endothelial cell activation by IL-1 and TNF-alpha stimulates the adhesion and transendothelial migration of circulating human CD14+ monocytes that develop with RANKL into functional osteoclasts. J Bone Miner Res. 2006;21:193-206.
69. Li Y, Horst K, Greven J, et al. Modulation of the hepatic RANK-RANKL-OPG axis by combined C5 and CD14 inhibition in a long-term polytrauma model. Front Immunol. 2024;15:1434274.
70. Moschen AR, Kaser A, Stadlmann S, et al. The RANKL/OPG system and bone mineral density in patients with chronic liver disease. J Hepatol. 2005;43:973-83.
71. Monti F, Perazza F, Leoni L, et al. RANK-RANKL-OPG axis in MASLD: current evidence linking bone and liver diseases and future perspectives. Int J Mol Sci. 2024;25:9193.
72. Raje NS, Bhatta S, Terpos E. Role of the RANK/RANKL pathway in multiple myeloma. Clin Cancer Res. 2019;25:12-20.
73. Buckle CH, De Leenheer E, Lawson MA, et al. Soluble rank ligand produced by myeloma cells causes generalised bone loss in multiple myeloma. PLoS ONE. 2012;7:e41127.
74. Chen G, Sircar K, Aprikian A, Potti A, Goltzman D, Rabbani SA. Expression of RANKL/RANK/OPG in primary and metastatic human prostate cancer as markers of disease stage and functional regulation. Cancer. 2006;107:289-98.
75. Wu X, Li F, Dang L, Liang C, Lu A, Zhang G. RANKL/RANK system-based mechanism for breast cancer bone metastasis and related therapeutic strategies. Front Cell Dev Biol. 2020;8:76.
76. Ohshiba T, Miyaura C, Inada M, Ito A. Role of RANKL-induced osteoclast formation and MMP-dependent matrix degradation in bone destruction by breast cancer metastasis. Br J Cancer. 2003;88:1318-26.
77. Honma M. [The potential of RANKL reverse signaling as a novel pharmacological target]. Nihon Yakurigaku Zasshi. 2023;158:253-7.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].