

The arsenic trioxide paradox: a targeted therapy triumph in APL versus a metabolic bottleneck beyond
Abstract
Arsenic trioxide (ATO) treatment has been successfully repurposed into a targeted therapy that achieves high cure rates in acute promyelocytic leukemia (APL) by degrading the PML-RARα oncoprotein (PML, promyelocytic leukemia protein; RARα, retinoic acid receptor alpha). Yet, its efficacy remains confined to APL, revealing a reliance on a precise molecular and permissive metabolic context not found in other cancers. Key biological barriers continue to limit its broader translation. To expand the therapeutic reach of arsenic-based agents, several critical challenges must be resolved. These include: (1) elucidating tumor-specific arsenic metabolic pathways that govern activation; (2) mapping comprehensive landscapes of intrinsic and adaptive resistance; (3) engineering tumor-selective prodrugs or delivery platforms to bypass metabolic dependencies; (4) identifying predictive biomarkers for patient stratification; and (5) rationally designing combination therapies to overcome multilayered resistance. Addressing these interconnected questions requires a concerted, interdisciplinary effort. By systematically dismantling these barriers, ATO can evolve from a remarkable single-disease therapy into a versatile and broadly applicable anticancer strategy.
Keywords: arsenic trioxide, acute promyelocytic leukemia, PML-RARα, drug resistance, drug metabolism
Highlights
1.Arsenic trioxide achieves curative efficacy in acute promyelocytic leukemia by targeting and degrading the PML-RARα fusion oncoprotein.
2.The success of arsenic trioxide in APL reflects a unique disease-specific molecular target that is rarely found in other cancers.
3.In non-APL malignancies, limited AS3MT-mediated activation and multilayered resistance mechanisms substantially restrict arsenic trioxide efficacy.
4.Precision strategies, including biomarker-guided stratification, tumor-selective prodrugs, efflux inhibition, and combination therapies, may broaden its anticancer application.
INTRODUCTION: THE APL-SPECIFIC PARADIGM AND THE CENTRAL PARADOX
Arsenic trioxide (ATO) treatment stands as a compelling testament to the transformative potential of molecularly targeted cancer therapy. ATO was originally known for its systemic toxicity. It has since been repurposed as a precision therapeutic agent for acute promyelocytic leukemia (APL), where it can induce deep and often curative remissions[1,2]. This success has solidified ATO’s status as a paradigm in oncology, demonstrating how a profound understanding of disease-specific molecular pathology can yield therapeutic breakthroughs.
However, the broader narrative of ATO is one of striking contrast. Attempts to extend ATO treatment to other hematologic malignancies or to solid tumors have yielded largely disappointing clinical outcomes[3]. This stark dichotomy between its efficacy in APL and its limitations elsewhere underscores a fundamental principle in cancer pharmacology. Therapeutic success is not merely a function of a drug's intrinsic cytotoxic potential, but is intricately contingent upon a permissive biological context [Figure 1].
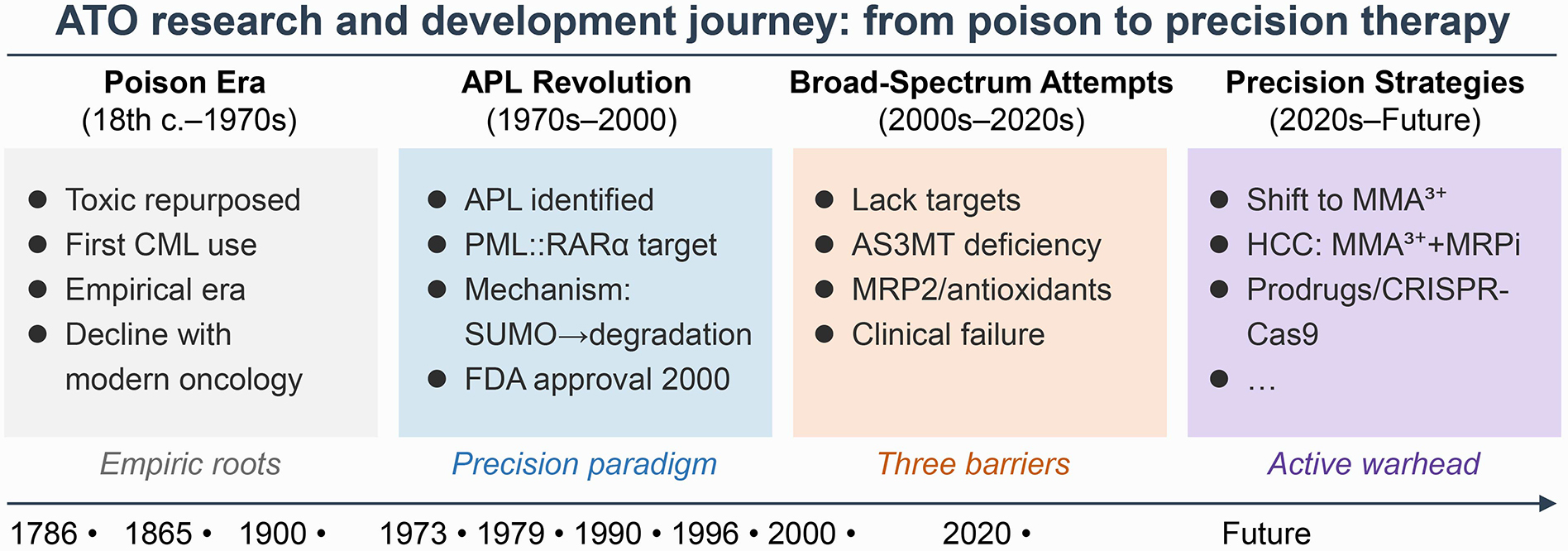
Figure 1. Arsenic trioxide (ATO) research and development journey: from poison to precision therapy.
The timeline depicts four distinct eras: (1) the Poison Era (18th century-1970s), characterized by empirical use based on observed toxicity without mechanistic understanding, followed by decline with the rise of modern oncology; (2) the APL Revolution (1970s-2000), marked by the identification of the promyelocytic leukemia protein (PML)-retinoic acid receptor alpha (RARα) fusion protein as a specific molecular target, elucidation of the small ubiquitin-related modifier (SUMO)ylation-dependent degradation mechanism, and subsequent Food and Drug Administration (FDA) approval, establishing a precision paradigm; (3) the Broad-Spectrum Attempts (2000s-2020s), which encountered clinical failures in non-APL malignancies due to the absence of analogous targets, deficiency of the metabolic activation enzyme AS3MT [arsenic(+3 oxidation state) methyltransferase], and robust efflux/antioxidant defenses; and (4) the ongoing Precision Strategies (2020s-future), which aim to overcome these barriers through innovative approaches including methylated arsenic species [e.g., monomethylarsonous acid (MMA3+)], prodrug design, efflux pump inhibition [e.g., multidrug resistance-associated protein (MRP) inhibitors], and functional genomics-guided combination therapies.
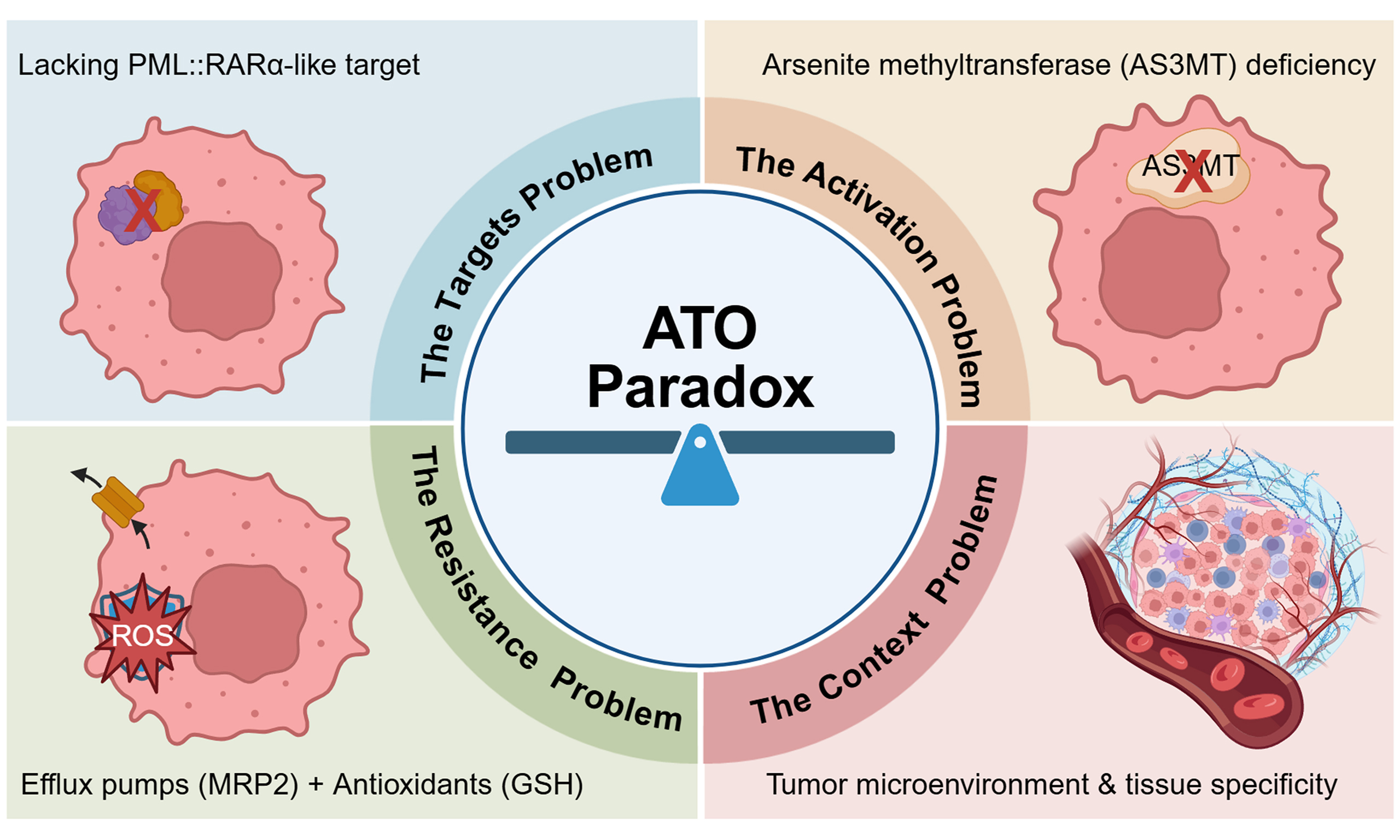
In APL, this context is uniquely favorable, encompassing three critical elements: (1) the presence of the uniquely targetable PML-RARα fusion protein, which arises from the t(15;17) chromosomal translocation specific to APL and absent in other cancer types, explaining the disease specificity of ATO treatment; (2) a metabolic landscape conducive to arsenic activation; and (3) a tumor cell intrinsically primed for differentiation and apoptosis upon oncoprotein degradation. Beyond APL, the absence of one or more of these conditions creates significant barriers. These barriers encompass not only the lack of an analogous high-affinity target but also profound challenges related to pharmacokinetics, tissue-specific metabolic activation, and pre-existing or adaptive resistance networks within tumor cells[4]. This perspective aims to dissect these multidimensional constraints that confine ATO's efficacy primarily to APL. More importantly, it seeks to chart a forward-looking, precision-guided roadmap to rationally overcome these barriers. A systematic analysis of the biological requirements for ATO activity may help redefine its therapeutic role. Combined with advances in chemical biology, functional genomics, and drug delivery, this approach could transform ATO treatment from a single-disease success into a broader therapeutic platform across multiple cancer types [Figure 2].
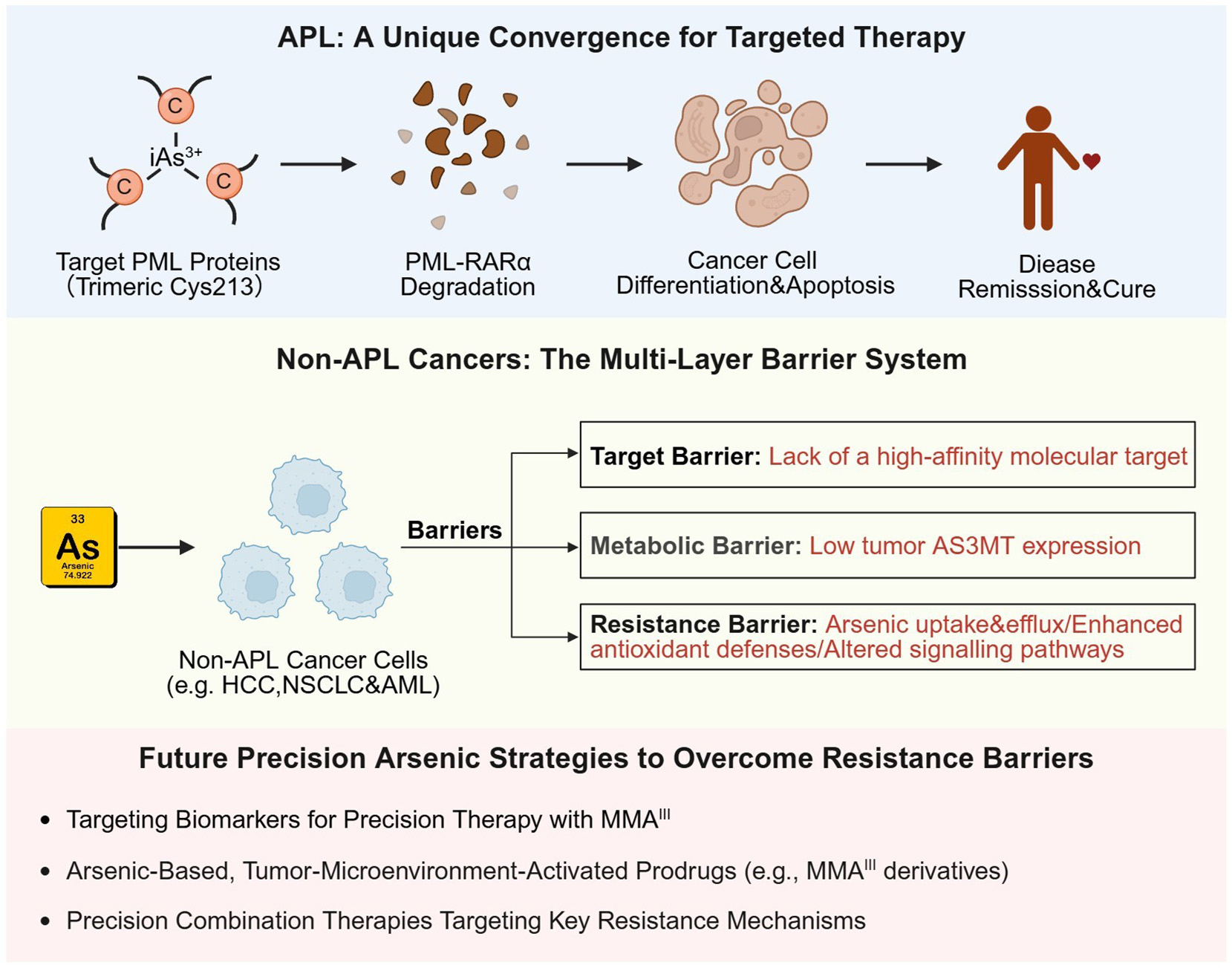
Figure 2. Mechanism of arsenic trioxide (ATO) in acute promyelocytic leukemia (APL) vs. multi-faceted barriers in non-APL cancers.
In APL, ATO can achieve curative outcomes by directly targeting the PML-RARα fusion oncoprotein. This interaction promotes degradation of the fusion protein and subsequently induces leukemic cell differentiation and apoptosis. In stark contrast, its efficacy in non-APL cancers is constrained by a lack of an analogous target and compounded by multiple systemic barriers. These include: (1) the absence of predictive biomarkers for patient stratification; (2) a metabolic bottleneck due to low expression of the activating enzyme AS3MT in extrahepatic tumors, limiting the local generation of active metabolites such as MMA3+; and (3) intrinsic and adaptive resistance mechanisms, including impaired cellular uptake, drug efflux transporters (e.g., MRP2), enhanced antioxidant defenses, and upregulated anti-apoptotic pathways. Overcoming these interconnected barriers will require a shift in development strategy. Rather than searching for a single “perfect” target, future efforts should focus on precision approaches that tailor drug activation, delivery, and combination therapy to the biology of each tumor type. Created in BioRender. Yang, C. (2026) https://BioRender.com/iewa6l8.
THE STRUCTURAL BASIS OF PRECISION TARGETING IN APL: A UNIQUE MOLECULAR LOCK AND KEY
The exceptional and selective efficacy of ATO treatment in APL is fundamentally rooted in its ability to perform a “precision strike” on a singular molecular entity: the PML-RARα fusion protein. This chimeric oncoprotein acts as a transcriptional repressor that blocks myeloid differentiation and drives leukemogenesis. ATO first binds to specific cysteine residues within the PML moiety of the fusion protein. This binding promotes multimerization and SUMOylation, followed by proteasomal degradation. Recent advances in structural biology have provided an atomic-resolution understanding of this exquisitely specific interaction. Studies reveal that ATO targets a sophisticated, condition-dependent trimeric interface formed by the B-Box2 domains (a cysteine-rich zinc-binding domain) of PML, coordinating with a triad of conserved cysteine residues within a central hydrophobic pocket[5]. This unique binding mode confers exceptionally high affinity and specificity for the PML-RARα oncoprotein.
The biological necessity of this precise interaction is powerfully validated in the clinic by the emergence of acquired resistance. Specific point mutations within the B2 domain can disrupt the architecture of this binding pocket or the trimeric interface, severely impairing ATO binding and rendering the leukemia refractory to treatment[6]. This structure-function relationship underscores that ATO treatment's success in APL relies on a unique trimeric arsenic-coordination pocket formed by cysteine residues in the B-Box2 domain. This scenario, however, represents a rare therapeutic alignment. The vast majority of other cancers lack an analogous, high-affinity protein target with a similar constellation of accessible cysteine residues arranged in a conducive spatial configuration. Consequently, the absence of a comparable “molecular lock” constitutes the primary and most fundamental barrier to translating the APL-like precision efficacy of ATO treatment to other malignancies. It shifts the challenge from one of pure target engagement to overcoming a series of downstream biological barriers that limit the activity of ATO’s core cytotoxic mechanisms.
METABOLIC BARRIERS: THE ACTIVATION DILEMMA AND TISSUE-SPECIFIC LIMITATIONS
In the treatment of APL, ATO primarily exerts its therapeutic effect through direct binding to PML-RARα and inducing its degradation, rather than relying on metabolic activation. However, in non-APL tumors, ATO faces a metabolic bottleneck that limits its efficacy, because a crucial aspect of ATO’s pharmacological effects is that the parent compound is not the ultimate cytotoxic effector. Its potent activity is largely dependent on biotransformation into even more reactive and toxic metabolites[7]. The key enzyme in this activation pathway is arsenic methyltransferase (AS3MT), which catalyzes the methylation of inorganic arsenic to form metabolites such as monomethylarsonous acid (MMAIII) and Dimethylarsinous acid (DMAIII). These methylated trivalent arsenicals are significantly more potent in inducing oxidative stress, disrupting mitochondrial function, and inhibiting various cellular enzymes[8].
The clinical challenge arises from the highly tissue-restricted expression pattern of AS3MT. In humans, AS3MT expression is predominantly enriched in the liver, with much lower levels found in most other tissues[9]. This creates a stark pharmacological disparity. For solid tumors originating from extrahepatic tissues, such as those of the breast, lung, prostate, or ovary, the tumor cells often reside in a “metabolic desert” with regard to arsenic activation. They largely lack the endogenous enzymatic machinery to efficiently convert administered ATO into its highly active methylated forms. Consequently, ATO may remain in a less active state even after reaching the tumor site. As a result, its intracellular concentration may not be sufficient to trigger substantial cytotoxic stress. This metabolic limitation provides a compelling explanation for the minimal single-agent activity observed in many solid tumor clinical trials[3].
INTRINSIC AND ADAPTIVE RESISTANCE: THE TUMOR'S MULTI-LAYERED DEFENSE NETWORK
Even in scenarios where ATO uptake and some degree of metabolic activation occur, tumor cells often deploy a formidable array of defensive countermeasures that confer intrinsic or adaptive resistance. This constitutes a third major barrier. Resistance to ATO treatment is rarely a single-gene event but is typically a systems-level adaptation involving multiple interconnected pathways[10,11].
Intrinsic resistance refers to pre-existing defenses present in tumor cells prior to ATO exposure, and common mechanisms include: (1) reduced cellular uptake by downregulation of aquaporin channels [i.e., aquaporin1-9 (AQP1-9)], preventing cytotoxic arsenic accumulation; (2) the upregulation of drug efflux pumps (e.g., MRP1-2), which can actively pump ATO and its metabolites out of the cell, reducing intracellular accumulation; (3) elevated expression of anti-apoptotic proteins, raising the threshold for apoptosis induction by arsenic-induced stress; (4) enhanced antioxidant machinery, especially glutathione synthesis; and (5) reprogramming of cellular energy metabolism to survive arsenic-induced mitochondrial stress[12,13].
Adaptive resistance to ATO develops under treatment-induced selective pressure, primarily through two mechanisms such as the selection of pre-existing resistant clones and the rewiring of key cellular signaling networks. For instance, ATO treatment often induces feedback activation of pro-survival pathways, allowing cells to endure and recover from the initial insult. These resistance mechanisms are not isolated; they interact synergistically with the metabolic barrier. The efficient efflux of ATO and its active metabolites decreases their intracellular half-life, thereby limiting their therapeutic action, while a strong antioxidant system neutralizes their reactive effects. Together, they construct a resilient “efficacy wall” that ATO must breach. Overcoming this wall requires moving beyond monotherapy and developing strategies to systematically disarm these cellular defenses.
TOWARD PRECISION APPROACHES: TUMOR-TYPE-SPECIFIC TRANSLATIONAL ROADMAPS
Given the heterogeneity of arsenic metabolism and resistance mechanisms across different cancers, a one-size-fits-all approach is destined to fail. The future of ATO treatment development lies in precision oncology strategies tailored to the distinct biological profile of each tumor type. This necessitates abandoning broad, empiric pan-tumor trials in favor of hypothesis-driven, biomarker-enriched studies.
Hepatocellular carcinoma (HCC) presents a unique yet complex opportunity because of its anatomical and metabolic context. As a liver-derived tumor, HCC cells reside in an environment with high basal AS3MT activity, which theoretically provides a “geographic metabolic advantage” for converting ATO into the more potent trivalent MMA3+[9]. However, this potential advantage is often counteracted in clinical practice, leading to suboptimal efficacy. A key reason lies in the robust excretory defense of hepatocytes and HCC cells: the MRP2 transporter efficiently pumps the newly formed MMA3+ out of the cells, preventing its cytotoxic accumulation[8]. This creates a paradoxical situation where high metabolic activation coexists with rapid efflux in some HCC, effectively neutralizing the therapeutic benefit of ATO. Therefore, this example underscores that merely increasing activation (e.g., via AS3MT) is insufficient. The translational strategy must evolve beyond single-target approaches to simultaneously address both activation and retention. A promising approach involves co-targeting this excretory pathway. Pharmacological inhibition of MRP2 represents a viable strategy to trap MMA3+ inside HCC cells. This forced intracellular retention, combined with MMA3+’s high affinity for critical cysteine residues in cellular enzymes, can effectively enhance its cytotoxic effects, transforming the metabolic potential into a clinical reality. This rationale directly supports exploring combination therapies with efflux pump inhibitors to block the escape of active metabolites, thereby sensitizing tumor cells.
In contrast, for the majority of solid tumors lacking endogenous AS3MT activity, the strategy must shift from utilizing to creating or bypassing the need for metabolic activation. This points to the realm of advanced drug design and delivery. A promising avenue is the development of smart prodrugs. The concept involves chemically synthesizing a stable, less toxic derivative of the ultimate effector, such as a protected form of MMA3+. This prodrug would be designed to remain inert during systemic circulation but incorporate a cleavable linker that is specifically activated by features abundant in the tumor microenvironment, such as overexpressed enzymes or acidic pH, enabling site-specific, precise toxin release[14]. Upon reaching the tumor, the prodrug would be selectively activated, releasing the potent payload locally and on-demand. This approach would completely decouple drug efficacy from the tumor's intrinsic metabolic capacity, transforming ATO from a metabolism-dependent agent into a context-responsive “intelligent” therapeutic.
SYSTEMATICALLY OVERCOMING RESISTANCE: MAPPING AND TARGETING THE DEFENSE NETWORK
A parallel and universally relevant strategy involves the systematic deconstruction of resistance landscapes. Instead of facing resistance as an empirical obstacle, modern functional genomics tools allow us to map it comprehensively. Using techniques such as genome-wide CRISPR-Cas9 [clustered regularly interspaced palindromic repeats (CRISPR)/Cas9] screens, researchers can identify genes whose loss or gain confers resistance or sensitivity to ATO in specific cancer models[15].
This knowledge enables the rational design of personalized combination regimens. The goal is to use a second agent to precisely target the identified “Achilles' heel” of the resistance network in a given tumor. For example, if functional genomics reveals that a tumor's resistance hinges on a specific antioxidant response pathway, combining ATO with an inhibitor of that pathway could be effective. Similarly, if reliance on certain anti-apoptotic proteins is key, adding agents that antagonize those proteins could restore apoptotic sensitivity. This approach moves beyond non-specific chemotherapy combinations towards mechanism-informed drug partnerships[16]. By pre-emptively dismantling the tumor's most critical defense nodes, we can resensitize it to the core cytotoxic mechanisms of arsenic.
CONCLUSION AND OUTLOOK
The journey of ATO treatment, from a feared poison to a curative agent in APL, and then to a drug facing significant challenges in broader oncology, encapsulates a profound lesson for cancer drug development. It shows that therapeutic efficacy is determined by multiple factors. These include the strength of target engagement, systemic pharmacokinetics, local metabolic activation, and the intrinsic and adaptive defense networks of tumor cells. ATO is highly effective in APL because several favorable conditions converge in this disease. These include a uniquely targetable oncoprotein, a biological context that supports its mechanism of action, and leukemic cells that are primed to undergo differentiation, as well as apoptosis after PML-RARα degradation.
To unlock the latent potential of ATO and related arsenic species beyond this singular indication, the research paradigm must evolve. The path forward lies in embracing precision translational science. This entails meticulously mapping the “arsenic-response profile” of different cancers, investing in innovative chemistry to develop next-generation, tumor-microenvironment-activated prodrugs, employing functional genomics to define tumor-specific resistance vulnerabilities, and developing robust biomarkers for patient stratification. By systematically deconstructing and innovatively overcoming the biological barriers that currently confine its utility, ATO can be transformed. Its future may not be as a simple, standalone drug for many cancers, but rather as the core component of a versatile therapeutic platform. This platform would integrate targeted delivery, controlled activation, and mechanism-based combination strategies tailored to the biological reality of each tumor type. In doing so, the legacy of ATO treatment would expand from being a remarkable historical case study to becoming an instructive model for the rational development of metallodrugs and the repurposing of complex biological agents in the era of precision oncology.
DECLARATIONS
Acknowledgments
We gratefully acknowledge the significant and rapid contributions of our colleagues to the field of arsenic research and regret that space limitations preclude a comprehensive discussion of all recent advances. The graphical abstract was created using BioRender (Yang, C., 2026; https://BioRender.com/tvody55).
Authors’ contributions
Contributed to discussions, writing, and critical revision of all sections of the manuscript: Zhu, C. Y.; Yi, R. H.; Yang, C.; Naranmandura, H.
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
Not applicable.
Financial support and sponsorship
This manuscript was funded by the National Natural Science Foundation of China (Nos. 825B2004, 82370191).
Conflicts of interest
Naranmandura, H. is an Editorial Board member of the journal Element. Naranmandura, H. was not involved in any steps of editorial processing, notably including reviewers' selection, manuscript handling or decision making. The other authors declare that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Shen, Z. X. et al. Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APL): II. Clinical efficacy and pharmacokinetics in relapsed patients. Blood 89, 3354-60 (1997).
2. Lo-Coco, F. et al. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N. Engl. J. Med. 369, 111-21 (2013).
3. Kumthekar, P. et al. A phase II trial of arsenic trioxide and temozolomide in combination with radiation therapy for patients with malignant gliomas. J. Neurooncol. 133, 589-94 (2017).
4. Balasundaram, N. et al. Metabolic adaptation drives arsenic trioxide resistance in acute promyelocytic leukemia. Blood Adv. 6, 652-63 (2022).
5. Bercier, P. et al. Structural basis of PML-RARA oncoprotein targeting by arsenic unravels a cysteine rheostat controlling PML body assembly and function. Cancer Discov. 13, 2548-65 (2023).
6. Yu, P. H.; Zhu, C. Y.; Kang, Y. Y.; Naranmandura, H.; Yang, C. Mutation in the unrearranged PML allele confers resistance to arsenic trioxide in acute promyelocytic leukemia. Research 8, 0696 (2025).
7. Hirano, S. Biotransformation of arsenic and toxicological implication of arsenic metabolites. Arch. Toxicol. 94, 2587-601 (2020).
8. Liu, Q. Toxicology of airborne inorganic arsenic: oxidative stress, molecular mechanisms, and organ-specific pathologies. Toxics 13, 753 (2025).
9. Qiu, T. et al. AS3MT facilitates NLRP3 inflammasome activation by m6A modification during arsenic-induced hepatic insulin resistance. Cell Biol. Toxicol. 39, 2165-81 (2023).
10. Ge, M. et al. Understanding and overcoming multidrug resistance in cancer. Nat. Rev. Clin. Oncol. 22, 760-80 (2025).
11. Tufail, M. et al. The hallmarks of oncogenic signaling: from pathways to resistance in cancer therapy. Drug Resist. Updat. 85, 101355 (2026).
12. Faubert, B.; Solmonson, A.; DeBerardinis, R. J. Metabolic reprogramming and cancer progression. Science 368, eaaw5473 (2020).
13. Reinfeld, B. I. et al. Cell-programmed nutrient partitioning in the tumour microenvironment. Nature 593, 282-8 (2021).
14. Shim, M. K.; Yang, S.; Sun, I. C.; Kim, K. Tumor-activated carrier-free prodrug nanoparticles for targeted cancer immunotherapy: preclinical evidence for safe and effective drug delivery. Adv. Drug Deliv. Rev. 183, 114177 (2022).
15. Guler Kara, H. et al. The G protein-coupled receptor GPR89A is a novel potential therapeutic target to overcome cisplatin resistance in NSCLC Calu1 cells. FEBS J. 292, 3755-70 (2025).
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
Article Notes
About This Article
Copyright





Data & Comments
Data
 0
0









Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].