

Asymmetrically coordinated single-atom catalysts: from synthetic strategy to structure-activity relationship
 0
0 Abstract
Asymmetric coordination structures in single-atom catalysts (SACs) represent a frontier in electrocatalysis, offering tunable electronic environments and enhanced catalytic performance beyond traditional symmetric M–N4 motifs. This review first categorizes asymmetric SACs into four structural families: (1) single-metal asymmetric coordination, achieved by heteroatom substitution or axial ligand incorporation; (2) non-contact multi-metal sites, where adjacent but unbonded metal atoms synergize electronically; (3) directly bimetallic-bonded asymmetric coordination structures; and (4) bridged multi-metal constructs connected via non-metal linkers (e.g., O, N, S). Key synthetic strategies, including metal–organic framework confinement, defect engineering, dual-solvent loading, and macrocyclic precursor mediation, are examined in detail. Then we summarize applications in oxygen reduction reaction and CO2 reduction reaction catalysis, and highlight how asymmetric coordination tunes intermediate adsorption energies, breaks scaling relations, and enables tandem catalysis to improve activity, selectivity, and stability. Advanced characterization techniques - aberration-corrected scanning transmission electron microscopy with electron energy loss spectroscopy, synchrotron X-ray absorption spectroscopy, and time-of-flight secondary ion mass spectrometry - are discussed for their roles in resolving atomic dispersion, coordination environment, oxidation states, and dynamic evolution under operando conditions. Finally, challenges and future directions are outlined, including precise low-temperature assembly of heteronuclear sites, scalability, long-term stability under harsh reaction conditions, selective pathway control, and the integration of operando analyses with theoretical modeling to guide rational catalyst design.
Keywords
INTRODUCTION
Fossil fuel extraction and utilization have triggered severe environmental problems, giving rise to an urgent global energy and environmental crisis. Efficient and clean energy storage and conversion technologies are essential for sustainable development, and, in the context of carbon peaking and carbon neutrality, mitigating CO2 emissions and promoting its valorization have emerged as critical objectives[1,2]. Electrochemical conversion of hydrogen energy represents a viable strategy for mitigating carbon dioxide emissions. Among the key reactions in hydrogen energy conversion, the oxygen reduction reaction (ORR) decisively influences the overall efficiency of hydrogen utilization[3-6]. In parallel, the electrochemical carbon dioxide reduction reaction (CO2RR), which converts CO2 into higher-value carbon-based products, such as CO, CH4, and HCOOH, holds tremendous promise for facilitating CO2 transformation[7-10]. It is noteworthy that nearly all energy conversion and storage technologies depend on high-efficiency catalysts[11-13]. Therefore, developing high-performance catalysts is essential for meeting sustainable development objectives. Although noble-metal catalysts exhibit superior intrinsic activity, their high cost and scarcity substantially impede large-scale practical deployment. Accordingly, designing catalysts that combine affordability with robust stability has become a critical priority in catalytic technology advancement[14].
Single-atom catalysts (SACs) have garnered extensive attention owing to their remarkable atomic utilization efficiency and distinctive structural features[15-17]. SACs consist of isolated metal atoms anchored on supports, offering atomically precise active centers, maximal metal utilization, and high structural tunability[18-20]. These features endow SACs with exceptional catalytic activity and selectivity. As a rapidly evolving class of supported metal catalysts, these catalysts are regarded as among the most promising materials for electrochemical applications[21-24].
In recent years, SACs with asymmetric coordination structures have gained considerable attention because of their unique properties and notable advancements[8]. Building on symmetrically coordinated SACs, researchers have successfully synthesized a range of SACs with asymmetric coordination structures by fine-tuning the types and ratios of metal precursors, coordinating atoms, and supports. These catalysts often exhibit outstanding catalytic performance and stability[25]. As an extension of symmetrically coordinated SACs, catalysts with asymmetric coordination mark a pivotal advancement. However, significant obstacles persist in synthesizing and characterizing atomic-site catalysts with asymmetric coordination structures. This review initially classifies asymmetrically coordinated SACs into two main categories: single-metal asymmetric coordination structures and multi-metal asymmetric coordination structures. Subsequently, the primary synthetic approaches for constructing asymmetrically coordinated SACs are detailed, and a summary of their ORR and CO2RR applications is provided. Finally, the key characterization techniques for these materials are succinctly discussed, and an outlook on the future development of asymmetric coordination structures is provided.
CONSTRUCTION AND STRUCTURAL CHARACTERISTICS OF ASYMMETRIC COORDINATION STRUCTURES
The M1–N4 configuration (where M is a metal atom and N is nitrogen) is a common active-center structure in single-atom electrocatalysts[26-29]. However, due to the relatively high electronegativity of the coordinated nitrogen atoms in the planar M1–N4 structure, the metal atom at the active center can bind reaction intermediates either too strongly or too weakly, resulting in sluggish catalytic kinetics and consequently hindering further improvements in electrocatalytic performance[30-33]. To address this issue, researchers have modulated the local coordination environment of the central metal atom by introducing heteroatoms (e.g., P, S, Cl[34-37]) to partially substitute for the coordinated nitrogen atoms, thereby constructing SACs with asymmetric coordination structures. This asymmetric coordination structure significantly modifies the electronic structure of the active center, effectively enhancing its catalytic activity during the reaction[38,39].
Catalysts with Multi-metal asymmetric coordination structures are atomic-level catalysts formed by introducing one or more metal atoms into a single-metal asymmetric coordination structure, thereby creating multiple metal sites or active centers with asymmetric coordination structures. These structures can be regarded as an extension of single-atom sites[40,41]. Catalysts with multi-metal asymmetric coordination architectures, building upon the advantages of single-atom (SA) catalysts, further optimize the adsorption states of reactants and intermediates, mitigate excessively strong or weak adsorption energies, disrupt linear scaling relationships among adsorbates, and enhance metal-atom loading[42-44]. Furthermore, the introduction of heterogeneous metals in multi-metal asymmetric coordination structures can effectively modify the electronic structure of the catalysts[41,45]. This modification involves regulating the charge state of active sites, altering electronic delocalization and orbital energy levels, and adjusting the d-orbital states of metal centers, which collectively optimize the electronic spin configuration. As a result, these changes influence the bonding and antibonding interactions with reactants, leading to improved adsorption and desorption processes[46,47]. In addition, for various reactions, synergistic interactions among the multiple metals can trigger in situ structural evolution under reaction conditions, thereby forming more favorable structural configurations. Consequently, catalysts with multi-metal asymmetric coordination structures exhibit excellent catalytic performance and significant potential in electrocatalysis[48-53].
In this chapter, we classify atomic-site catalysts with asymmetric coordination structures into two major categories: single-atom asymmetric structures and multi-metal asymmetric coordination structures. Based on the morphological characteristics of multi-metal asymmetric coordination structures, these catalysts can be further divided into non-contact multi-metal, directly bimetallic-bonded, and bridged multi-metal asymmetric coordination structures. The main strategies for synthesizing these distinct types of catalysts and their corresponding structural features are discussed individually.
Single-metal asymmetric coordination structures
Single-metal asymmetric coordination structures are constructed on the basis of the conventional M–N4 coordination motif by introducing additional heteroatoms either within the M–N4 equatorial plane or along the axial direction of the active center, and by tuning the coordination number of the active site. Such modifications can modulate the charge distribution at the catalytic site, the d-band center, and the adsorption state, thereby enhancing catalytic activity. Employing this strategy to improve the overall performance of SACs has emerged as a highly effective and broadly applicable approach.
Based on this, Wan et al. encapsulated triphenylphosphine (PPh3) within a Zn/Co bimetallic metal–organic framework (MOF) cage and pyrolyzed it under argon at 950 °C to obtain a Co-SA/P catalyst bearing
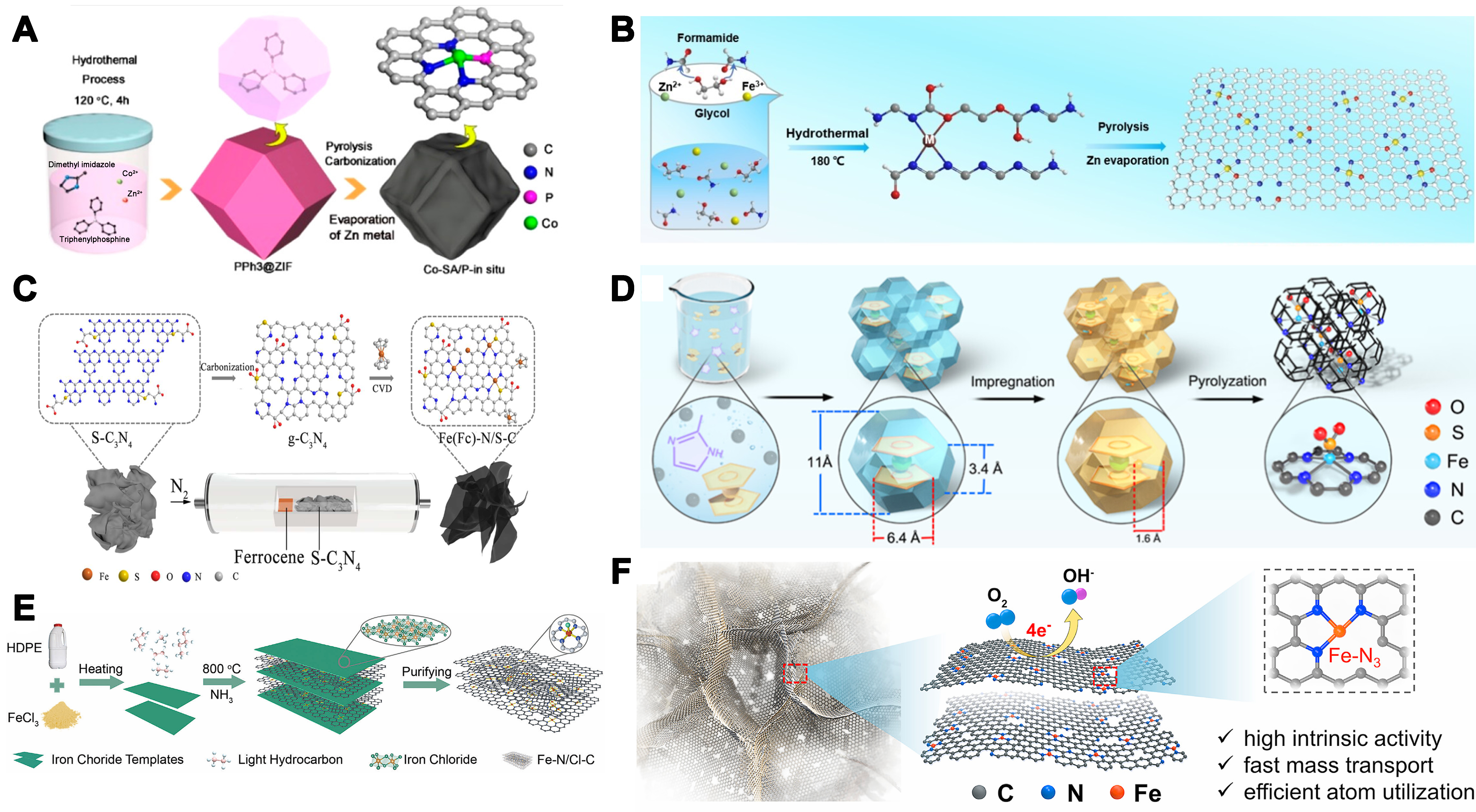
Figure 1. (A) Synthesis and morphological characterizations of Co-SA/P-in situ[54]. Copyright 2020, American Chemical Society; (B) Schematic diagram of synthesized Fe-N/O-C[55]. Copyright 2024, Wiley-VCH; (C) Typical CVD procedures to synthesize the Fe(Fc)-N/S-C catalyst[56]. Copyright 2021, American Chemical Society; (D) Schematic illustration of the synthesis of the Fe1-N4SO2/NC catalyst[57]. Copyright 2025, American Chemical Society; (E) Schematic illustration and characterization of the Fe-N/Cl-C catalyst synthesized from HDPE[58]. Copyright 2025, Wiley-VCH; (F) Schematic diagram of the synthesis route for SACs-Mn-1000@g-C3N4[60]. Copyright 2021, Cell Press. SA: Single-atom; CVD: chemical vapor deposition; HDPE: high-density polyethylene; SACs: single-atom catalysts.
Furthermore, introducing axially coordinated non-metal atoms into the traditional M–N4 structures, as well as adjusting the central metal’s coordination number to form M–N4-x structures, are both key approaches to constructing single-atom asymmetric coordination architectures. A brief overview of their synthesis methods is provided below.
In the preparation of catalysts with axial coordination, Qu et al. employed potassium thiocyanate (KSCN) as the sulfur-oxygen dopant, impregnated it onto a FeCp2@ZIF-8 precursor via ultrasonication and stirring in methanol, and then pyrolyzed the dried composite under N2 at 950 °C for 3 h to yield Fe1-N4SO2/NC featuring atomically dispersed Fe1-N4SO2 coordination sites on a carbon matrix [Figure 1D][57]. In addition, Ren et al. impregnated high-density polyethylene (HDPE) with FeCl3, pyrolyzed under NH3 at 800 °C for
Non-contact multi-metal asymmetric coordination structures
Non-contact coordination structures differ from randomly dispersed single-atom structures. In non-contact coordination structures, although the metal atoms do not directly contact each other, the spacing between them needs to be sufficiently small (< 15 Å). Two metal atoms with sufficiently small spacing can interact to achieve the desired catalytic performance. Therefore, non-contact multi-metal asymmetric coordination structures have attracted extensive attention from researchers.
As reported by Zhang et al., a quasi-double-star Ni7/Fe3-N-C catalyst was produced via one-pot solvothermal incorporation of Ni and Fe into Zn-IRMOF-3, followed by Ar-atmosphere pyrolysis at 950 °C for 2 h, yielding a carbon-supported material with atomically dispersed adjacent Ni and Fe SA sites[62]. Han et al. encapsulated Pt(acac)2 and Fe(acac)3 in ZIF-8 and obtained Fe-N4/Pt-N4@NC by pyrolyzing the precursor[63]. Density functional theory (DFT) calculations showed that adjacent Pt-N4 sites can effectively modulate the 3d electronic orbitals of Fe-N4 sites, resulting in enhanced catalytic performance [Figure 2A]. Building on this concept of adjacent-site electronic modulation, Zhao et al. developed a low-temperature pyrolysis of diethylenetriaminepentaacetic-acid bimetallic complexes to produce dynamically stable carbon dots with embedded bimetallic atomic sites (DMASs-CDs)[64]. As shown in Figure 2B, more than twenty DMASs-CDs incorporating Fe, Co, Ni, Mn, Zn, Cu, and Mo pairings were prepared. Notably, NiMn-CDs exhibited superior intrinsic activity in the urea oxidation probe reaction. By adjusting DMAS combinations, this method enables precise, target-oriented design of catalysts for various electrochemical processes. Remarkably, non-noble transition metals such as Co, Cu and Ni also furnish exceptional catalytically active sites. As shown in Figure 2C, the bifunctional oxygen electrocatalyst (Cu–Co/NC), comprising copper–cobalt dual-atom sites with optimized geometric and electronic structures on a high-porosity nitrogen-doped carbon (NC) support, was prepared by Li et al.[65]. Specifically, after introducing copper acetate into Zn-Co MOF, they coated it with a polymer rich in nitrogen sources and then pyrolyzed it in an inert atmosphere at high temperatures to obtain the target catalyst. Experimental and theoretical results showed that Cu and Co atoms exist as atom pairs on the carbon-nitrogen support. Both Cu and Co formed M-N4 coordination structures, with Cu in a +1 oxidation state and Co in a +2 oxidation state. After loading Cu and Co, the electronic properties of N and C changed, with N predominantly existing in the form of metal-pyridinic N. Similary, Wu et al. synthesized SOD-[Zn(mim)2] (MAF-4/ZIF-8, Hmim = 2-methylimidazole) by modulating the Zn/Co/Ni feed ratios during MOF synthesis under vigorous stirring[66]. Under an N2 atmosphere, the MOF precursors were combined with NH4Cl and pyrolyzed at 900 °C to yield carbonized samples. Inductively coupled plasma mass spectrometry (ICP-MS) quantification established the Co and Ni contents, and the products were designated AD-Co1Ni0, AD-Co0.72Ni0.28, AD-Co0.62Ni0.38, AD-Co0.48Ni0.52, AD-Co0.41Ni0.59, AD-Co0Ni1, and NC-Co0.49Ni0.51. Powder X-ray diffraction (PXRD) and high-resolution transmission electron microscopy (HRTEM) confirmed the absence of nanocrystals or nanoparticles/clusters. High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) imaging revealed numerous bright spots on the carbon matrix, attributable to atomically dispersed Co and Ni atoms, many separated by distances less than 5.0 Å, suggesting the coexistence of multiple bimetallic site configurations [Figure 2D].
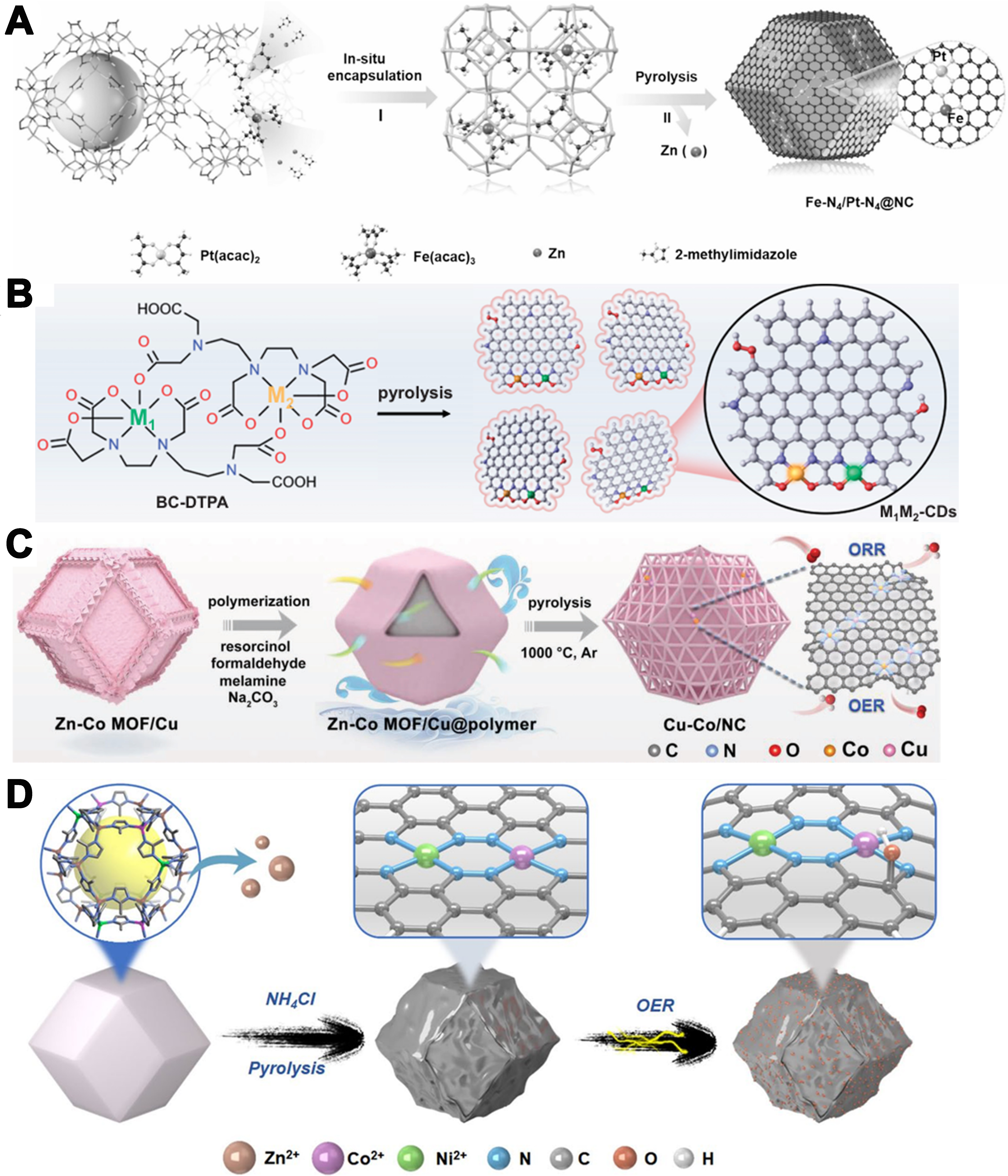
Figure 2. (A) Synthesis process of Fe-N4/Pt-N4@NC[63]. Copyright 2021, Wiley-VCH; (B) Preparation and structure illustration of the “carbon islands” of DMASs-CDs (red: O atom, blue: N atom, gray: C atom, green: first metal atom, orange: second metal atom)[64]. Copyright 2023, National Academy of Sciences; (C) Synthetic scheme of Cu-Co/NC[65]. Copyright 2023, Wiley-VCH; (D) Schematic illustration of the synthetic processes and structures of AD-CoxNi1-x and O-AD-CoxNi1-x[66]. Copyright 2023, Springer Nature. DMASs-CDs: Dynamically stable carbon dots with embedded bimetallic atomic sites.
Directly bimetallic-bonded asymmetric coordination structures
Direct bimetallic-bonded asymmetric coordination structures refer to catalysts that contain two (or more) different or identical single-atom active sites, where these single-metal atoms are bonded to each other. Unlike metal alloys, these directly bonded metals can only be anchored into the substrate in single-atom form. Such interactions between metals often effectively optimize and regulate the electronic structure, coordination environment, electron spin configuration, and charge state of the active sites.
As shown in Figure 3A, Zhang et al. synthesized the Zn/Co/Ni imidazole zeolite framework (Zn/Co/Ni-ZIFs) precursor using cation exchange and cavity adsorption strategies, and finely tuned the structure of the target catalyst NiN3©CoN3-NC by adjusting the Zn content and pyrolysis temperature[67]. Through aberration-corrected HAADF-STEM, 3D atomic overlap Gaussian fitting mapping, X-ray absorption fine structure (XAFS), and X-ray diffraction (XRD), they systematically confirmed the structural characteristics of NiN3©CoN3-NC (Ni-Co covalently coupled atom pair embedded in nitrogen-doped graphitized carbon). Moreover, Wang et al. synthesized C2N supports featuring uniform N6 cavities and edge N deficiencies by high-temperature pyrolysis of cyclohexanone and urea in the presence of MgCl2, providing optimal sites for metal anchoring[68]. Fe and Sn precursors were subsequently introduced via wet impregnation followed by a second pyrolysis, yielding the FeSn-C2N dual-atom catalyst with Fe and Sn atoms precisely anchored within the N6 cavities, forming a robust structure. Energy band alignment between Sn p-orbitals and Fe d-orbitals enhanced p-d coupling and improved the catalyst’s thermodynamic stability, underscoring its catalytic potential. XAFS characterization revealed that Fe in FeSn-C2N exhibited a lower oxidation state than in Fe-C2N, due to the polarization-charge effects induced by Sn incorporation. Fe K-edge FT-EXAFS confirmed Fe–Sn bonding at 2.22 Å, verifying the bimetallic coordination [Figure 3B]. Sn K-edge X-ray absorption near edge structure (XANES) indicated a reduced Sn oxidation state relative to Sn-C2N, reflecting charge redistribution from strong Fe-Sn electronic interactions. In summary, the reduced oxidation states of Fe and Sn in FeSn-C2N reflect the combined effect of Fe-Sn bimetallic electronic coupling and polarization charge redistribution.
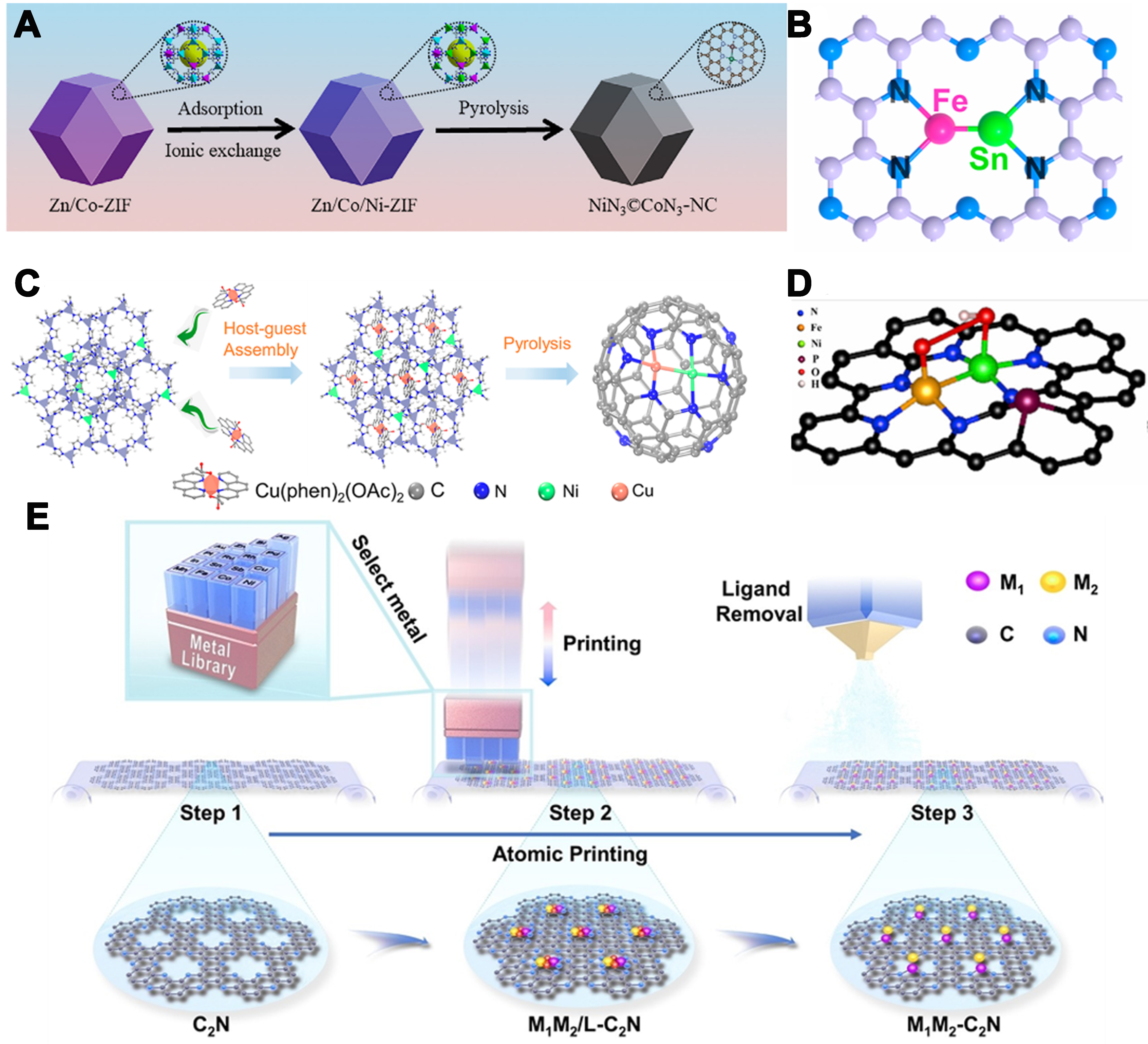
Figure 3. (A) Scheme illustration of the synthetic procedure of NiN3@CoN3-NC[67]. Copyright 2023, Elsevier B.V; (B) The dimer structure of active FeSn dual atom sites derived from the EXAFS result[68]. Copyright 2024, American Chemical Society; (C) Schematic illustration of the synthetic procedure of the diatomic Ni/Cu-N-C catalyst[69]. Copyright 2022, American Chemical Society; (D) The geometric structure of Fe-Ni-N-P-C[70]. Copyright 2021, Cell Press; (E) Schematic illustration of the synthesis of the Cu2-C2N[71]. Copyright 2024, Wiley-VCH. EXAFS: Extended X-ray absorption fine structure.
Furthermore, Zhu et al. reported a Ni–Cu atomic pair catalyst (Ni/Cu-N-C) synthesized via a host–guest strategy [Figure 3C][69]. A Cu/1,10-phenanthroline complex (~9.5 Å) was encapsulated within a bimetallic NiZn zeolitic imidazolate framework (Ni–ZIF-8; cavity ~11.6 Å), ensuring isolated Cu sites in the ZIF micropores. Zn dilution and pore confinement favored the formation of Ni–Cu pairs over nanoparticles or single atoms. Characterization showed no metal nanoparticles and revealed a Ni–Cu interatomic distance of ≈2.4 Å, indicating atomically dispersed metals with strong electronic coupling. To achieve highly active SACs, Pan et al., guided by DFT and Monte Carlo (MC) simulation calculations, used a dual-solvent method to load Fe3+ and Ni2+ onto ZIF-8 as a template, followed by co-pyrolysis with sodium hypophosphite to obtain P-doped Fe-Ni dual-atom pair catalysts (Fe-Ni-N-P-C)[70]. Both theoretical and experimental results successfully demonstrated the precise synthesis of the P-doped Fe-Ni dual-atom pair [Figure 3D].
As shown in Figure 3E, the facile atomic printing strategy to fabricate a series of M1M2-C2N materials was developed by Sun et al.[71]. In a typical synthesis, a solid mixture of cyclohexanone and urea was heated at
Bridged multi-metal asymmetric coordination structures
In Bridged multi-metal asymmetric coordination structures, there is no direct interaction between the metal atoms, but they are connected by non-metallic atoms (such as O, N, S). Under the influence of non-metallic atoms, the electronic density between bimetallic sites can be redistributed, which alters the charge state of the metals and enhances catalytic performance. Non-metallic bridging atoms with different electronegativities often result in completely different catalytic effects.
Based on this, Zhang et al. prepared planar-Fe-Co and stereo-Fe-Co dual-site catalysts (DSCs) through various electrochemical deposition methods[73]. Experimental characterizations revealed the different spatial relationships between Fe single atoms and Co atoms in CoOOH substrates, confirming that the Fe and Co atoms in the catalysts exist in an Fe-O-Co form. Furthermore, Zhao et al. developed a dual-atom catalyst (DAC) synthesis approach at the simple-level stage by exploiting precise electrostatic control and engineered adjacent vacancies[74]. As a proof of concept, they fabricated uniformly dispersed dual-iron sites bridged by two nitrogen atoms (Fe-N2-Fe). Their method anchors a second Fe ion at pre-existing SAC sites using bridging nitrogen precursors; subsequent pyrolysis etches neighboring carbon atoms to form vacancies that trap the second ion [Figure 4A]. Similarly, Sun et al. noted that incorporating sulfur ligands into SACs typically requires high-temperature pyrolysis to convert solid sulfur into gaseous species for substrate etching, a process that usually yields only monometal–sulfur coordination at bimetallic sites, precluding sulfur-bridged architectures[75]. To address this, they employed wool-derived keratin as a support to construct a dual-metal catalyst featuring novel Cu–S–Ni bridging sites (Cu–S–Ni/SNC). The abundant disulfide bonds in keratin serve as in situ sulfur donors, while the protein’s partial long-range order provides defined anchoring sites for metal atoms. Additionally, X-ray absorption spectroscopy (XAS) elucidated the coordination geometry of the Cu–S–Ni sulfur-bridged bimetallic sites [Figure 4B].
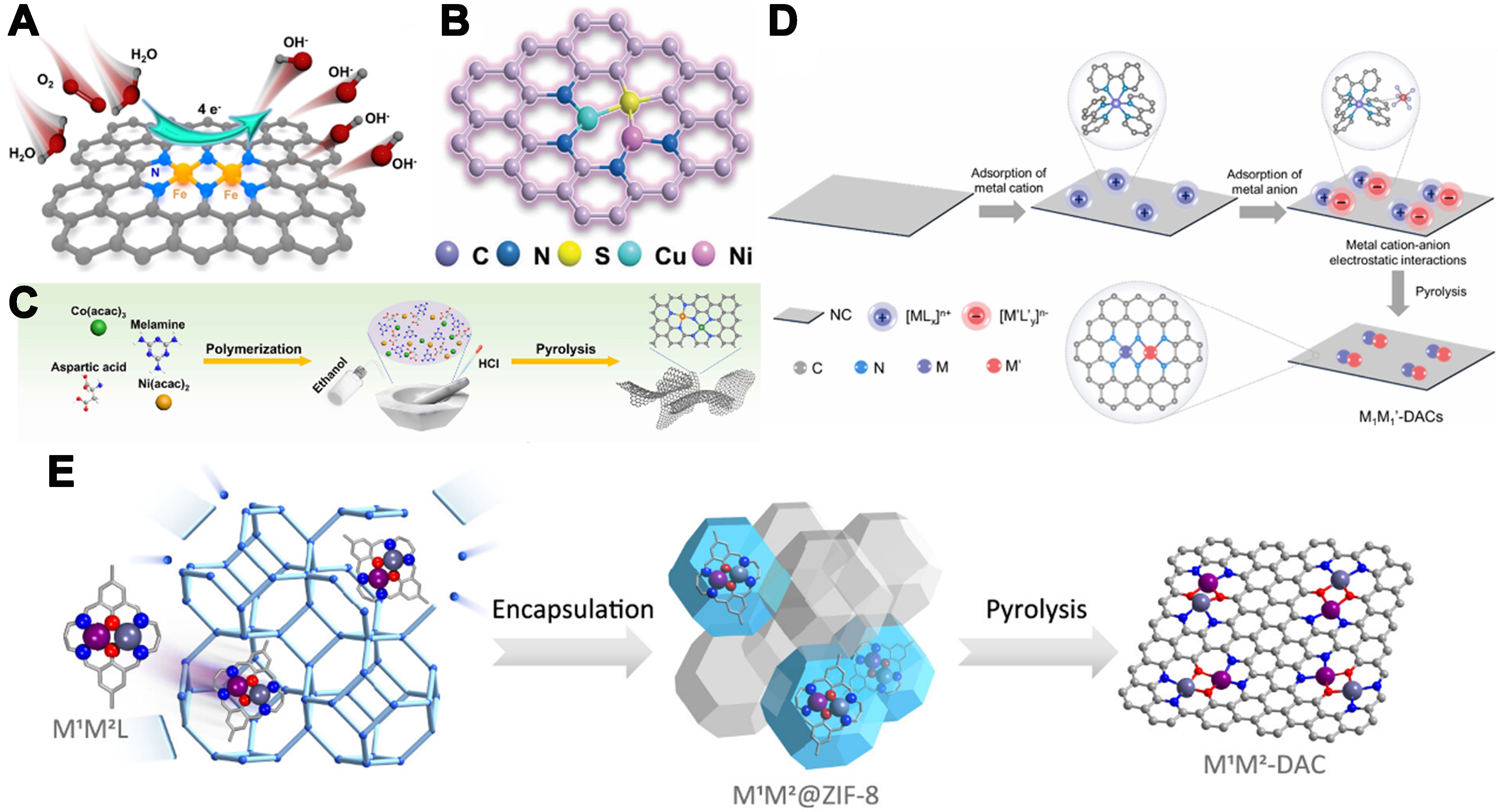
Figure 4. (A) Atomic structure model of Fe-N2-Fe DAC[74]. Copyright 2024, Wiley-VCH; (B) Atomic structure model of Cu-S-Ni/SNC[75]. Copyright 2024, Wiley-VCH; (C) Schematic diagram of the polymerization-pyrolysis synthesis method of Co-N-Ni[76]. Copyright 2023, American Chemical Society; (D) Schematic Illustration of the MIR strategy for DACs[77]. Copyright 2024, American Chemical Society; (E) Schematic illustration of the synthesis of DACs via a macrocyclic precursor-mediated encapsulation-pyrolysis process. The model on the far right represents the local structure of the M1M2-DAC[78]. Copyright 2023, American Chemical Society. DAC: Dual-atom catalyst; SNC: sulfur- and nitrogen-co-doped carbon; MIR: metal ion recognition.
Li et al. prepared a Co-N-Ni catalyst by co-grinding melamine, L-aspartic acid, Co(acac)3, and Ni(acac)2; adding ethanol/HCl and evaporating the solvent; then pyrolyzing under N2 and cooling[76]. Characterization confirmed that this polymerization–decomposition route precisely controls the N-bridged Co-Ni spacing [Figure 4C]. As shown in Figure 4D, Wang et al. developed an innovative metal ion recognition (MIR) strategy to address the challenges in the synthesis of dual-atom catalysts[77]. This strategy involves the sequential adsorption of target cations and anions onto NC substrates to form precursors, enabling the synthesis of various DACs. Taking Fe1Sn1-DAC as an example, NC derived from ZIF-8 with rhombic dodecahedral morphology and negative surface charge was first prepared. Then, [Fe(bpy)3]2+ and [SnCl6]2- were sequentially adsorbed to form a heterometallic bimetallic ion pair, followed by high-temperature pyrolysis to obtain Fe1Sn1-DAC. Multiple characterization techniques confirmed the successful synthesis of this catalyst, and theoretical calculations revealed the coordination structure of Fe and Sn. Additionally, other DACs such as Fe1Co1, Fe1Ni1, Fe1Cu1, Fe1Mn1, Co1Ni1, Co1Cu1, Co2, and Cu2 were also successfully synthesized, demonstrating the versatility of the strategy. Similarly, Zhang et al. introduced a macrocycle-mediated encapsulation–pyrolysis strategy for precise DAC synthesis[78]. A Robson-type macrocycle embeds homo- and heteronuclear bimetallic centers within a planar ligand framework, and its confinement in porous carbon preserves the dual-atom motif during heat treatment. Employing this method, a Fe–Cu dual-atom site bridged by two oxygen atoms was constructed [Figure 4E].
APPLICATIONS OF ASYMMETRIC COORDINATION STRUCTURES IN ELECTROCHEMICAL REACTIONS
Single-metal asymmetric coordination structures
The coordination environment of the central metal in SACs has a crucial impact on catalytic performance. Studies have shown that incorporating the central atom with one or more coordination atoms of different electronegativity can significantly enhance the catalyst’s catalytic performance.
Recent in-depth studies on SACs have pointed out that the symmetric M-N4 structure of traditional SACs, due to its symmetric charge state, is not conducive to the adsorption-desorption and catalytic conversion of all intermediates in catalytic reactions. This results in high reaction barriers and low catalytic selectivity for M-N4 structure catalysts. Therefore, replacing one of the nitrogen atoms in M-N4 with other atoms of different electronegativity can impart strong polarity and a special charge state to the metal center, thereby improving its catalytic activity.
Proton exchange membrane fuel cells (PEMFCs) and zinc–air batteries efficiently convert chemical energy into electricity, offering significant promise for new energy vehicles and flexible electronics. However, the sluggish kinetics of the ORR limits their electrocatalytic efficiency. SACs have gained widespread attention due to their fully exposed active sites and high atomic utilization. Current research mainly focuses on symmetric M-N4 sites anchored on carbon-nitrogen materials. The symmetric M-N4 sites, due to their uniform electronic distribution, have relatively limited adsorption capability for different intermediates during the ORR process. In contrast, single-atom sites with asymmetric coordination structures exhibit asymmetric electronic distributions, and through effective site regulation, can enhance the adsorption and activation of various intermediate species.
Recently, Li et al. synthesized an Fe-N/O-C catalyst featuring isolated Fe–N3O sites adjacent to vacancy defects[55]. By introducing O into the first coordination shell and coupling with nearby defects, they tuned the Fe d-orbital energy and spin state from low to intermediate spin, allowing the unpaired dz2 electrons to interact with O π* orbitals to mitigate over-binding [Figure 5A]. The catalyst achieves ORR half-wave potentials of 0.927 V in 0.1 M KOH and 0.80 V in 0.1 M HClO4 [Figure 5B and C], and zinc–air and PEMFC peak power densities of 490 and 1,179 mW·cm-2, respectively [Figure 5D]. DFT shows a more negative Fe d-band center in Fe–N/O–C vs. Fe–N–C, weakening intermediate adsorption and shifting the rate-determining step from OH* desorption to OOH* formation. In addition, A coal-pitch-derived Fe SAs/N and S co-doped carbon (NSC)-vacancy defects (vd) catalyst was synthesized via supramolecular self-assembly pyrolysis, yielding asymmetric Fe–N3S1 single-atom sites stabilized by abundant vacancy defects[79]. This vacancy-induced Fe–N3S1 coordination synergistically enhances ORR kinetics, delivering markedly improved turnover frequency (TOF) and mass activity in both alkaline and acidic media, far surpassing 20 wt% Pt/C [Figure 5E and F]. Mechanistic analysis indicates that Fe–N sites predominantly facilitate O2 adsorption activation under alkaline conditions, whereas Fe–S sites govern ORR in acidic media. DFT calculations confirm that the elevated d-band center of Fe–N3S1 sites promotes O2 adsorption and OH reduction, while vacancy defects balance *OOH formation and *OH reduction to collectively drive the electrocatalytic ORR. Low-electronegativity boron atoms were introduced by Guan et al. to construct an asymmetric Co–N3B SAC[80]. Relative to Co-N4, Co-N3B reduces ORR free energy and strengthens *O adsorption, thereby suppressing the two-electron pathway and H2O2-induced corrosion to enhance stability. In situ attenuated total reflection (ATR)-surface-enhanced infrared absorption spectroscopy (SEIRAS) confirmed robust Co–O intermediate interactions [Figure 5G]. In rechargeable Zn–air batteries, Co-N3B delivers ≈253 mW·cm-2 peak power,
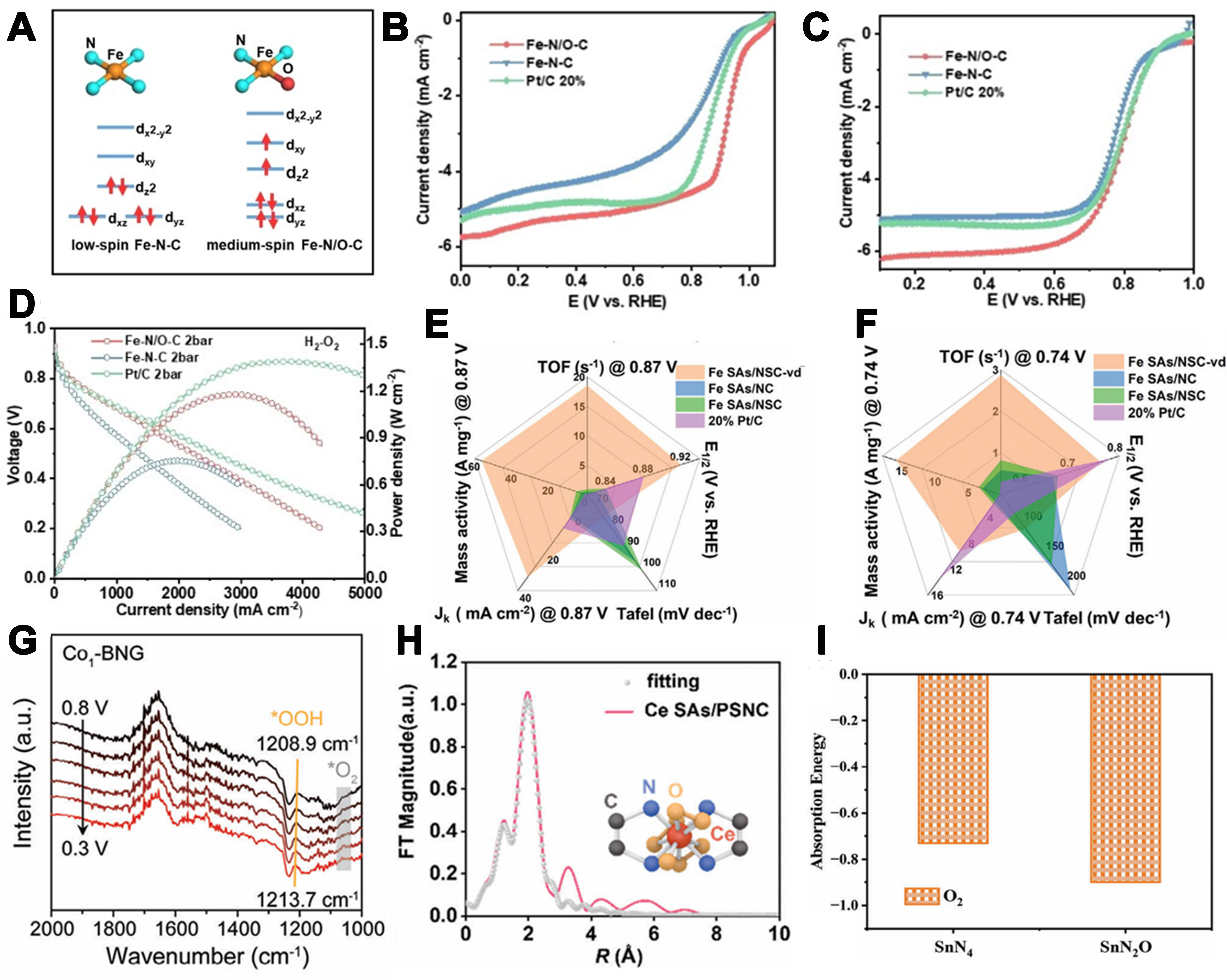
Figure 5. (A) Schematic diagram of d-orbital spin-electron filling states; (B) LSV curves of FeN/OC, FeNC and Pt/C 20% in 0.1 M KOH at the rotation speed of 1,600 rpm; (C) LSV curves of FeN/OC, FeNC and Pt/C 20% in 0.1 M HClO4 at the rotation speed of 1,600 rpm; (D) Polarization curves and corresponding power density plots of FeN/OC, FeNC and Pt/C-assembled 2.0 bar H2-O2 PEMFCs[55]. Copyright 2024, Wiley-VCH; (E) E1/2, Jk, Tafel, mass activity, and TOF at 0.87 V of the catalysts in 0.1 M KOH and (F) 0.5 M H2SO4[79]. Copyright 2024, Wiley-VCH; (G) In situ ATR-SEIRAS spectra of ORR over Co1/BNG in O2-saturated 0.1 M KOH[80]. Copyright 2024, Wiley-VCH; (H) The corresponding EXAFS R-space fitting curves of CoSAs/PSNC[81]. Copyright 2023, Wiley-VCH; (I) O2 absorption energies on the optimized model structures of SnN4 and SnN2O[83]. Copyright 2024, Wiley-VCH. LSV: Linear sweep voltammetry; PEMFCs: proton exchange membrane fuel cells; TOF: turnover frequency; ATR-SEIRAS: attenuated total reflection-surface-enhanced infrared absorption spectroscopy; ORR: oxygen reduction reaction; EXAFS: extended X-ray absorption fine structure.
Electrochemical CO2 reduction into fuels or value-added chemicals offers a promising route to mitigate energy and environmental challenges, garnering extensive interest from both academia and industry. Nevertheless, sluggish CO2 activation kinetics and the scarcity of efficient electrocatalysts necessitate high energy inputs, impeding industrial deployment. Consequently, designing electrocatalysts with enhanced activity and selectivity is critical. Transition-metal SACs supported on N-doped porous carbon have demonstrated considerable advancement in CO2 electro conversion, yet their symmetric atomic coordination constrains catalytic performance. Precisely tuning the atomic-level coordination environment to optimize intermediate adsorption and facilitate charge transfer to the single-atom active sites remains a formidable challenge. Recently, Huang et al. employed Mg(OH)2 as a low-cost template, whose decomposition during pyrolysis generated water vapor for axial Ni-O coordination, while ammonia doping formed lateral Ni-N4 sites on porous graphitic carbon[84]. The resulting Ni SA-N-PGC catalyst, featuring asymmetric Ni-N4-O coordination, achieved over 97% CO Faradaic efficiency (FE) at -0.76 V (9,097 h-1) and maintained > 90% efficiency across -0.5 to -1.1 V, with negligible decay after 40 h. Mechanistic studies revealed that axial Ni–O coordination induced site asymmetry, optimized the local coordination environment, enhanced charge polarization, and lowered the Gibbs free energy of *COOH formation, thereby improving ORR kinetics and selectivity [Figure 6A and B]. Jin et al. designed an asymmetric
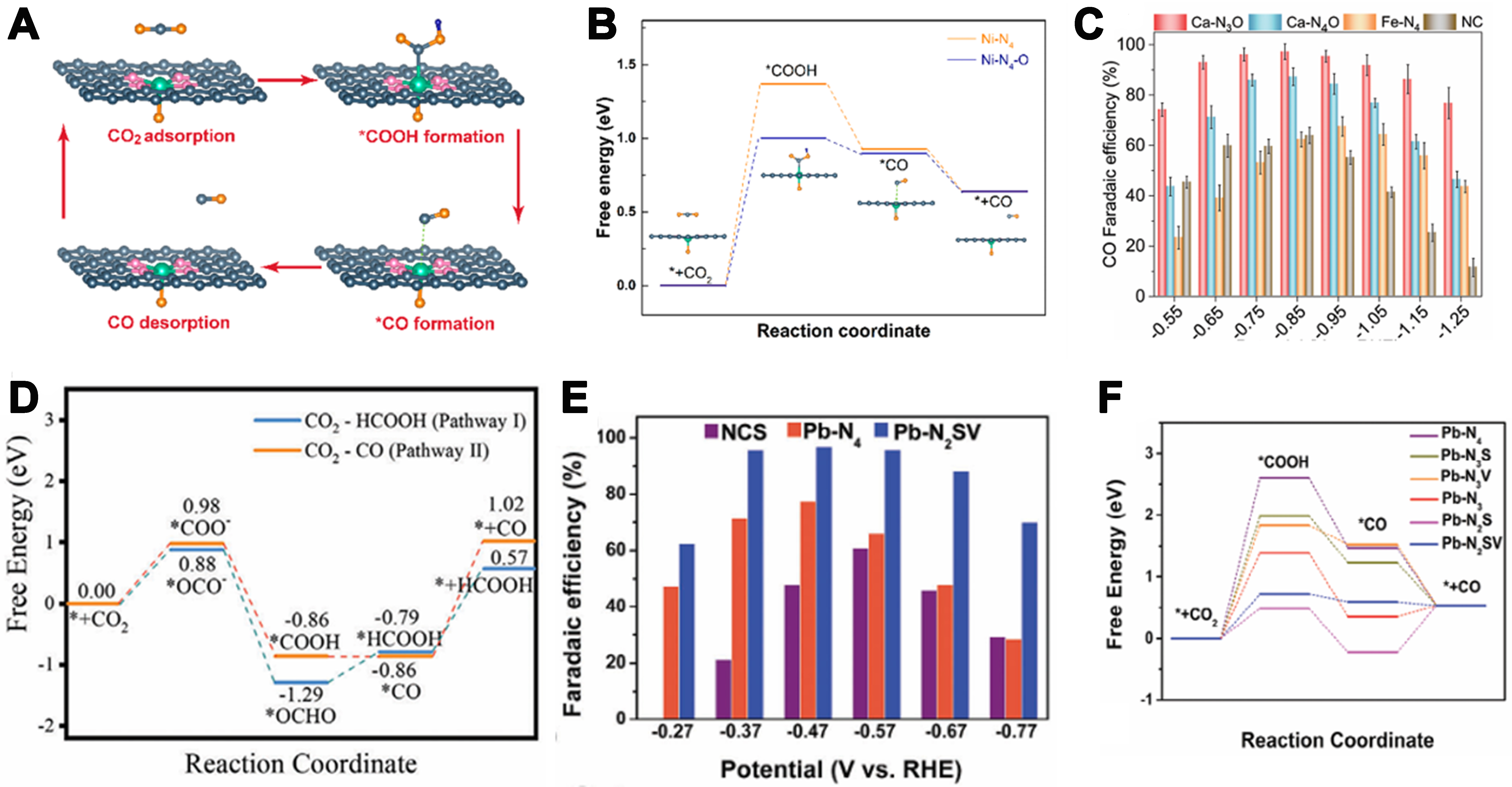
Figure 6. (A) Proposed reaction steps of electrocatalytic reduction of CO2 to CO over the Ni-N4-O catalytic site; (B) Reaction paths and free energy diagrams of CO2 reduction to CO over Ni-N4 and Ni-N4-O catalytic sites. Inset shows the side view of the optimized configurations of intermediates over the Ni-N4-O catalytic site[84]. Copyright 2022, American Chemical Society; (C) FECO at different potentials in H-cell[86]. Copyright 2023, Wiley-VCH; (D) The Gibbs free energy diagrams of pathways I (from CO2 to HCOOH) and II (from CO2 to CO)[87]. Copyright 2024, Wiley-VCH; (E) Comparison of FECO at different potentials for NSC, Pb-N4 and Pb-N2SV in CO2-saturated 0.5 M KHCO3; (F) Gibbs free energy profiles[88]. Copyright 2024, Wiley-VCH. FECO: CO Faradaic efficiency; NSC: sulfur- and nitrogen-co-doped carbon.
Multi-metal asymmetric coordination structures
In complex multistep reactions, the adsorption and desorption strengths of various intermediates at the active sites are often different. However, SACs feature only a single active center and therefore exhibit the same adsorption/desorption response for all intermediates. Therefore, multi-metal asymmetric coordination structures have been introduced into electrochemical systems with multiple reaction intermediates (e.g., ORR, OER, NRR, CO2RR[40,67,68,73]), enabling differentiated control over each intermediate’s adsorption and desorption behavior. Multi-metal asymmetric coordination structures can effectively regulate the charge state, orbital energy levels, d-band center, optimize electronic spin configurations, and influence the bonding and antibonding states of reactants, thus optimizing the adsorption and desorption of intermediates during the reaction. Moreover, multi-metal sites enable tandem catalysis during the reaction process. Different active site centers can achieve optimal adsorption and desorption capabilities for various intermediates, allowing the reaction to proceed smoothly[89-91]. In this section, we will discuss the applications of multi-metal asymmetric coordination structures in ORR and
As shown in Figure 7A, Li et al. synthesized a Cu-Co/NC catalyst with outstanding bifunctional ORR/OER activity in alkaline media (ORR E1/2 = 0.92 V), and maintained strong ORR performance in acidic (0.85 V) and neutral (0.74 V) electrolytes[65]. When applied in zinc–air batteries, it delivered stable open-circuit voltage, polarization, and power density, and endured 1,000 charge-discharge cycles (510 h at 10 mA·cm-2) without performance loss. DFT analysis revealed that Cu-Co dual sites induce asymmetric charge distribution, optimizing adsorption/desorption of oxygen intermediates and accelerating both ORR and OER kinetics. It is noteworthy that diatomic catalysts composed of two identical metal atoms likewise exhibit high catalytic activity. Li et al. synthesized a charge-asymmetric dual-atom catalyst Fe2-S1N5/SNC using a two-step method[92]. Fe2-S1N5/SNC exhibited excellent electrocatalytic performance in acidic flow cells. In 0.1 mol·L-1 HClO4 solution, its half-wave potential reached 0.829 V, an improvement of 56 mV compared to the SAC Fe-N4/NC. When loaded on the cathode of a hydrogen fuel cell, it demonstrated a high peak power density of 810 mW·cm-2 [Figure 7B]. The authors attributed the catalyst’s excellent performance to the fact that charge-asymmetric dual-atom sites can more flexibly regulate the adsorption energy of point-charged intermediates at the active center, and the asymmetric electronic structure enhances the electronic transfer capability. In 2024, He et al. synthesized Co-Fe-SNC via a host–guest strategy to create bridging asymmetric Co-Fe coordination[93]. Atomically dispersed Co-Fe sites, combined with sulfur-driven surface modulation, deliver superior ORR performance, achieving a half-wave potential of 0.92 V in alkaline media [Figure 7C]. DFT studies reveal that bimetallic sites strengthen oxygen-intermediate binding and lower activation barriers, while sulfur’s weak electronegativity fine-tunes the electronic structure for further activity enhancement. Employed as zinc–air battery cathodes, Co-Fe-SNC surpasses commercial Pt/C in power density, specific capacity, and durability. It is worth noting that Li et al. engineered a Fe-Ru dual-atom catalyst on N-doped carbon with moderate tensile strain, achieving record acidic ORR performance among Fe SACs[94]. Strain-tuned d-band center yielded a half-wave potential of 0.86 V and a fuel-cell power density of 700 mW·cm-2. After 50,000 cycles, the half-wave potential dropped by only 17 mV, demonstrating exceptional durability. In situ SEIRAS and DFT analyses reveal that tensile strain enhances d-electron penetration into antibonding orbitals, accelerating intermediate reduction and desorption. ATR-SEIRAS further confirmed strain-optimized Fe-O bonding and faster ORR kinetics in acid.
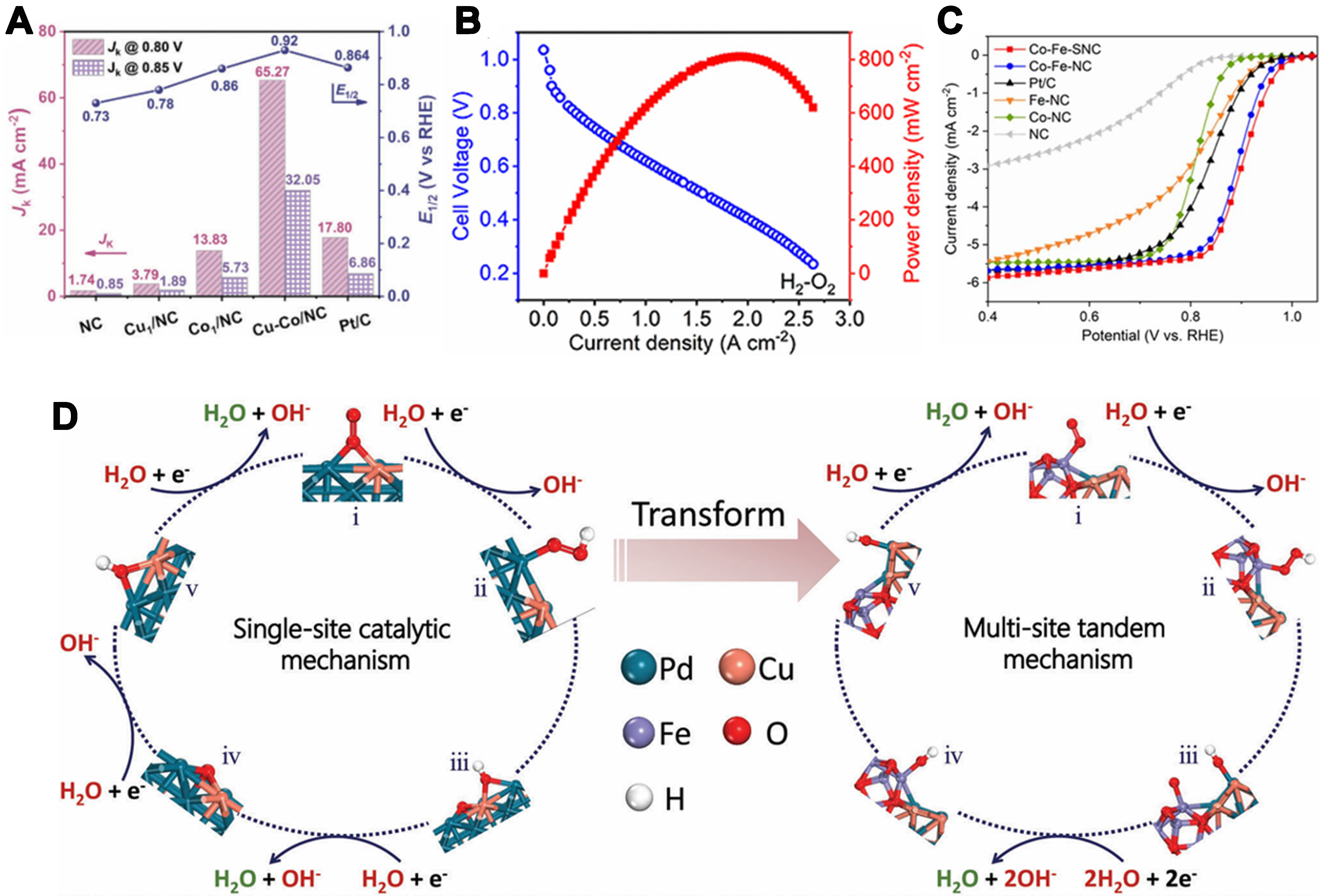
Figure 7. (A) Kinetic current density of electrocatalysts at 0.80 and 0.85 V[65]. Copyright 2023, Wiley-VCH; (B) PEMFC polarization and power density curves of Fe2-S1N5N5/SNC[92]. Copyright 2024, The Royal Society of Chemistry; (C) LSV polarization curves of the ORR in O2-saturated 0.1 M KOH solution[93]. Copyright 2024, The Royal Society of Chemistry; (D) Schematic illustration of the single-site catalytic mechanism for PdCu (left) and multi-site tandem mechanism for PdCu-Fe3O4 (right)[95]. Copyright 2024, Wiley-VCH. PEMFC: Proton exchange membrane fuel cell; LSV: linear sweep voltammetry; ORR: oxygen reduction reaction.
Furthermore, asymmetric catalysts utilizing metal oxides as supports have likewise demonstrated superior ORR performance. For example, Li et al. successfully synthesized a PdCu-Fe3O4 in-plane heterostructure through sequential reduction of different metal precursors[95]. This structure combines the advantages of Pd and Fe3O4, achieving synergistic catalysis. The authors pointed out that this method ensures the uniformity and stability of the heterostructure. ORR catalytic performance evaluation of the prepared PdCu-Fe3O4 in-plane heterostructure showed that it maintained close to 4e- electron transfer numbers at different potentials, indicating its high catalytic activity. Notably, the study found that the proton-coupled electron transfer (PCET) steps during ORR occur in an orderly manner at Pd and Fe sites. The initial PCET steps occur at Fe sites, while the last two PCET steps take place at the optimal Pd sites [Figure 7D]. This reveals the tandem catalysis mechanism of ORR on the PdCu-Fe3O4 in-plane heterostructure, providing a new research paradigm for understanding ORR mechanisms in complex systems.
Remarkably, catalysts featuring multi-metal asymmetric coordination structures also find extensive applications in the CO2RR. For example, Zhu et al. constructed a quasi-covalently bonded Ni–Cu dual-atom electrocatalyst (Ni/Cu-N-C) exhibiting exceptional CO2 reduction performance[69]. It delivers peak current densities and the lowest onset potential across the full potential window, achieving 13.7 mA·cm-2 at -0.7 V
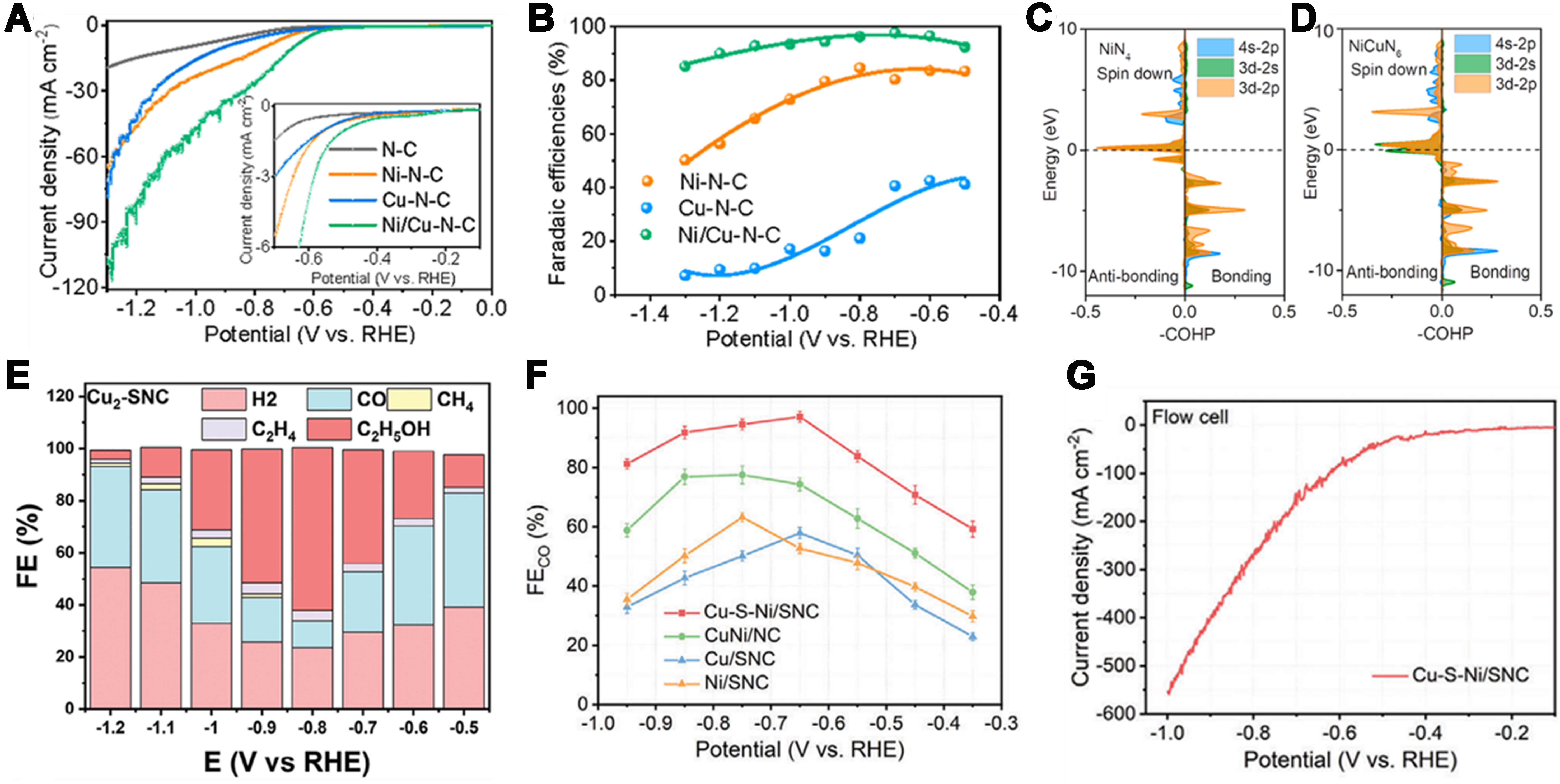
Figure 8. (A) LSV curves acquired in CO2-saturated 0.5 M KHCO3 solution on a rotating disc electrode at a rotating speed of 1,600 rpm. The inset highlights the LSV curves in the potential range from -0.1 to -0.7 V; (B) FE for CO production at various applied potentials; (C and D) COHP analysis of the Ni–C bond on NiN4 and NiCuN6; note that -COHP is used as the a-axis[69]. Copyright 2022, American Chemical Society; (E) FE value at different potentials by Cu2-SNC catalysts[96]. Copyright 2024, Wiley-VCH; (F) FECO at different potentials of Cu-S-Ni/SNC and references; (G) LSV curves of Cu-S-Ni/SNC in the flow cell[75]. Copyright 2024, Wiley-VCH. LSV: Linear sweep voltammetry; FE: Faradaic efficiency; COHP: crystal orbital Hamilton population; SNC: sulfur- and nitrogen-co-doped carbon.
CHARACTERIZATION METHODS FOR ASYMMETRIC COORDINATION STRUCTURES
In the rapidly expanding domain of multi-scale design for single-atom catalysis, a thorough grasp of the subtle structural characteristics inherent in catalytic materials is essential for achieving exceptional catalytic performance. At the nanoscale and below, the intricate organization of atoms and molecules plays a fundamental role in determining the catalytic activity, selectivity, and longevity of SACs. To decipher these intricate structural, morphological, and electronic properties, advanced characterization techniques are indispensable. This section outlines key advanced characterization techniques: aberration-corrected STEM coupled with electron energy-loss spectroscopy (EELS), XAS, and time-of-flight secondary ion mass spectrometry (ToF-SIMS).
Aberration-corrected scanning transmission electron microscopy with electron energy loss spectroscopy
Electron microscopy techniques, including scanning electron microscopy (SEM), transmission electron microscopy (TEM), and HRTEM, have been widely used for morphology characterization of catalysts. However, they are limited by insufficient resolution and thus cannot accurately characterize individual atoms. To address this limitation, aberration-corrected scanning transmission electron microscopy (AC-STEM) has been developed to observe single atoms. In particular, spherical aberration-corrected HAADF-STEM (AC-HAADF-STEM) can display larger metal atoms as prominent bright spots. This allows for the observation of single-atom sites and dual-atom sites in catalyst materials. Sun et al. used AC-HAADF-STEM to characterize the Cu-S-Ni/SNC catalyst they prepared, as shown in Figure 9A, where the yellow circles highlight isolated bimetallic sites, and the red circles encircle individual Cu or Ni single atoms[75]. Figure 9B shows the 3D model of the dual-atom site at position 1 in Figure 9A. The authors statistically analyzed most of the sites in the image, and the results showed that bimetallic sites accounted for more than 70% [Figure 9C], confirming that Cu-Ni bimetallic sites dominate in the catalyst. Similarly, Xie et al. used AC-HAADF-STEM to confirm that most of the Zn-Fe metals in their prepared Fe-Zn@SNC catalyst exist as isolated dual-atom sites (as shown in the yellow circle in Figure 9D)[97]. Furthermore, the authors statistically analyzed the distance between the dual-atom sites, and the results Figure 9E showed that the distance between dual-atom sites in Fe-Zn@SNC catalyst is primarily concentrated between 2.8 to 3.3 Å.
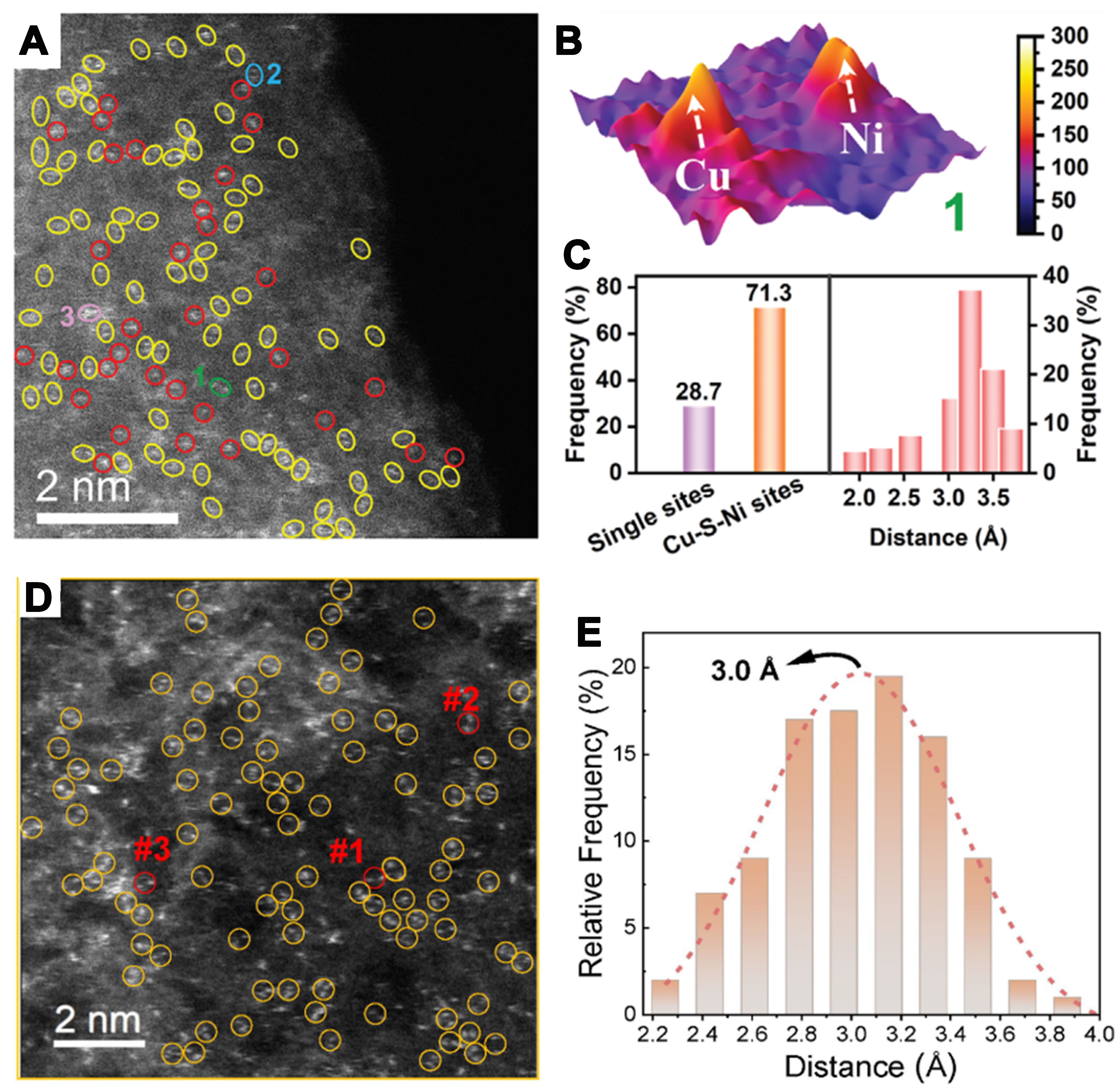
Figure 9. (A) HAADF-STEM image of Cu-S-Ni/SNC. The dual-atom sites and single sites were circled with dotted yellow line and red line, respectively; (B) 3D model of Cu-S-Ni sites along the region of 1 in (A); (C) Density profiles of Cu-S-Ni and single-atom sites in the HAADF-STEM[75]. Copyright 2024, Wiley-VCH; (D) HAADF-STEM image of Fe-Zn@SNC; (E) Kernel method of frequency statistic and Gauss fitting of diatomic distances obtained from all the yellow circles in (D)[97]. Copyright 2023, Wiley-VCH. HAADF-STEM: High-angle annular dark-field scanning transmission electron microscopy; SNC: sulfur- and nitrogen-co-doped carbon.
However, AC-STEM cannot provide information on the chemical composition and elemental identification of the sample, which is complemented by EELS. In recent years, EELS has offered unparalleled advantages for the characterization of SACs and DACs. By measuring the energy-loss spectrum of a high-energy electron beam transmitted through a specimen, EELS yields rich information about elemental composition, chemical bonding, oxidation state and coordination environment[98]. When combined with aberration-corrected high-resolution scanning transmission electron microscopy (STEM), it enables sub-angstrom localization of individual atoms, while the energy-loss spectrum confirms elemental identity and discriminates impurity atoms. This structural–chemical dual-criterion approach permits both qualitative and quantitative determination of atomic positions and oxidation states, thereby effectively eliminating misidentification[99-101]. The STEM–EELS tandem thus provides significant advantages in active-site identification of SA catalysts, elemental qualitative–quantitative analysis, microstructural characterization and in situ dynamic observation. Zhang et al. used HAADF-STEM to characterize the FeCu bimetallic catalyst they prepared (the schematic structure of which is shown in Figure 10A)[78]. Figure 10B shows a large number of bright spots dispersed on the carbon support, which are considered to be metal sites. The researchers pointed out that the proportion of metal atom pairs is about 70%. Additionally, the authors performed EELS analysis on the atomic pair within the red box in Figure 10B, and the results, shown in Figure 10C, confirmed that the two bright spots within the red box are Fe-Cu atomic pairs. As shown in Figure 10D, Yang et al. undertook a two-phase method to create Cu-N-C SACs, featuring a uniform and distinctly defined Cu2+-N4 configuration[102]. It is also confirmed the dispersion of Cu atoms by observing HAADF-STEM images [Figure 10E], and the clear signals of C, N, and Cu in EELS, combined with elemental mapping, further corroborated the N/C coordination [Figure 10F]. Despite the single-atom sensitivity of AC-STEM with EELS and its capability to obtain rich physicochemical properties, the requirement for sample stability under the electron beam necessitates the combination of other characterization techniques for analysis[98].
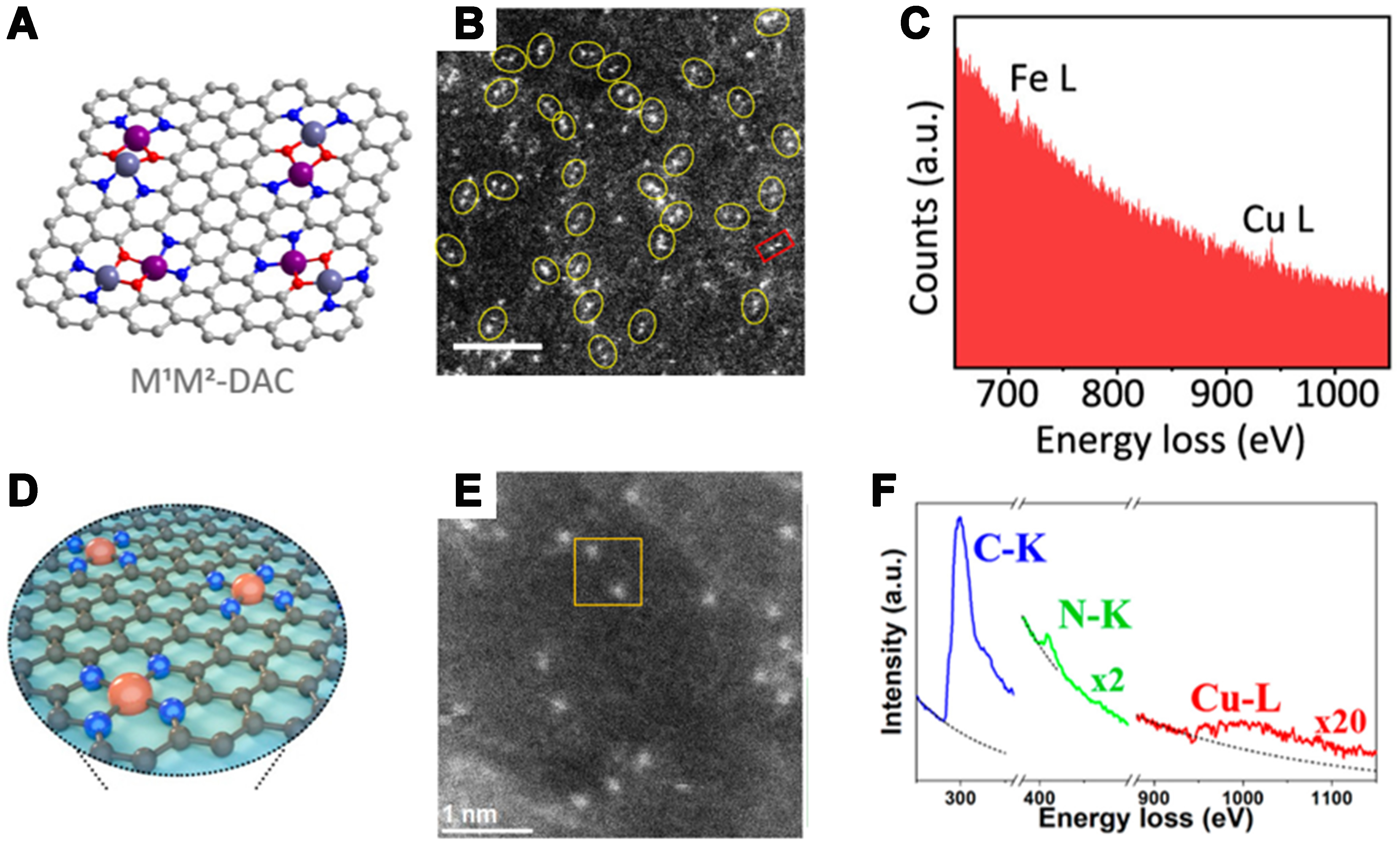
Figure 10. (A) The local structure of the M1M2-DAC; (B) HAADF-STEM image of FeCu-DAC; (C) EELS of an Fe-Cu atomic pair as marked in the red rectangle in the HAADF-STEM image[78]. Copyright 2023, American Chemical Society; (D) The structure of Cu-N-C SAC; (E) Atomic-resolution STEM-HAADF image; (F) The EELS extracted from the yellow rectangle in STEM-HAADF image[102]. Copyright 2021, American Chemical Society. DAC: Dual-atom catalyst; HAADF-STEM: high-angle annular dark-field scanning transmission electron microscopy; EELS: electron energy-loss spectroscopy; SAC: single-atom catalyst.
XAS
Synchrotron radiation XAS plays a crucial role in the study of asymmetric coordination structures. XAS includes extended XANES and extended XAFS (EXAFS). XANES can provide information on the atomic electronic orbitals and oxidation states in SACs, as well as the electron density distribution, oxidation state changes, and the localization of electrons in single atoms. This is particularly important for analyzing the electron transfer processes in catalytic reactions. On the other hand, EXAFS can reveal the local structure of single atoms in detail, including coordination environment, bond lengths, and coordination numbers, which can help researchers understand the arrangement of surrounding atoms in SACs. Furthermore, by utilizing the high temporal resolution capabilities of synchrotron radiation XAS, researchers can monitor the dynamic changes of SACs in real-time during catalytic reactions, capturing the evolution of their structure and electronic states at different stages of the reaction[103-107]. Additionally, the high temporal resolution of synchrotron radiation XAS allows researchers to monitor the dynamic changes of SACs during catalytic reactions in real time, capturing the evolution of their structure and electronic states at different stages of the reaction.
As shown in Figure 11A, Pei et al. investigated the local coordination environment of Co-SxN4-x SACs using XANES and FT-EXAFS techniques[108]. The XANES spectra revealed that, compared to Co foil and Co3O4, the Co in SACs carries a positive charge and exhibits a lower valence state, which decreases as the S content increases. FT-EXAFS analysis [Figure 11B] confirmed the single-atom structure of Co and unveiled a new Co-S scattering peak that intensifies with increasing S content. Through EXAFS fitting [Figure 11C-F], researchers extracted structural parameters, finding that an increase in Co-S coordination number leads to a decrease in Co-N coordination number, indicating the substitution of N by S.
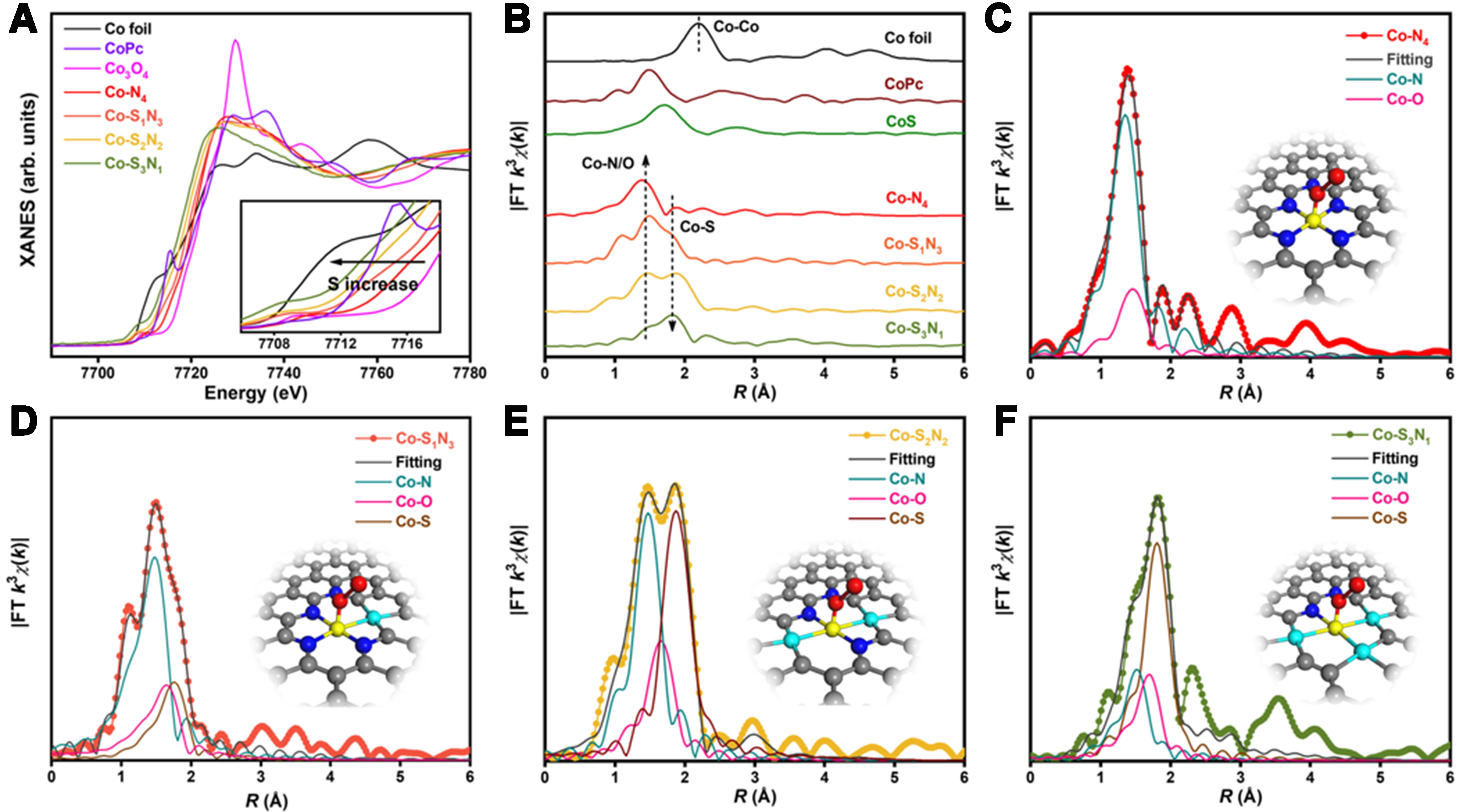
Figure 11. (A) XANES and (B) FT-EXAFS spectra of the Co-SxN4-x SACs; (C) Co-N4, (D) Co-S1N3, (E) Co-S2N2 and (F) Co-S3N1[108]. Copyright 2021, Springer Nature. XANES: X-ray absorption near edge structure; FT-EXAFS: Fourier transform extended X-ray absorption fine structure; SACs: single-atom catalysts.
Similarly, Sun et al. studied the local coordination structure of the Cu-S-Ni asymmetric sites in Cu-S-Ni/SNC catalysts using XANES and EXAFS techniques [Figure 12A][75]. Figure 12B and C shows the normalized Cu and Ni K-edge XANES spectra of Cu-S-Ni/SNC and comparative samples. The results show that Cu in Cu-S-Ni/SNC has a higher oxidation state compared to Cu foil, presenting a positive oxidation state, but its oxidation state lies between that of CuNi/NC and Cu/SNC, indicating that the oxidation state of Cu is influenced by Ni and S elements. Similarly, the Ni K-edge XANES spectrum shows that Ni in Cu-S-Ni/SNC also exhibits a positive oxidation state, with its oxidation state between that of CuNi/NC and Ni/SNC, which is mainly due to the effect of the coordination environment and the doping elements [Figure 12C]. These observations suggest that the electronic oxidation states of Cu and Ni are both between 0 and +2, and are influenced by the S atoms and the second metal.
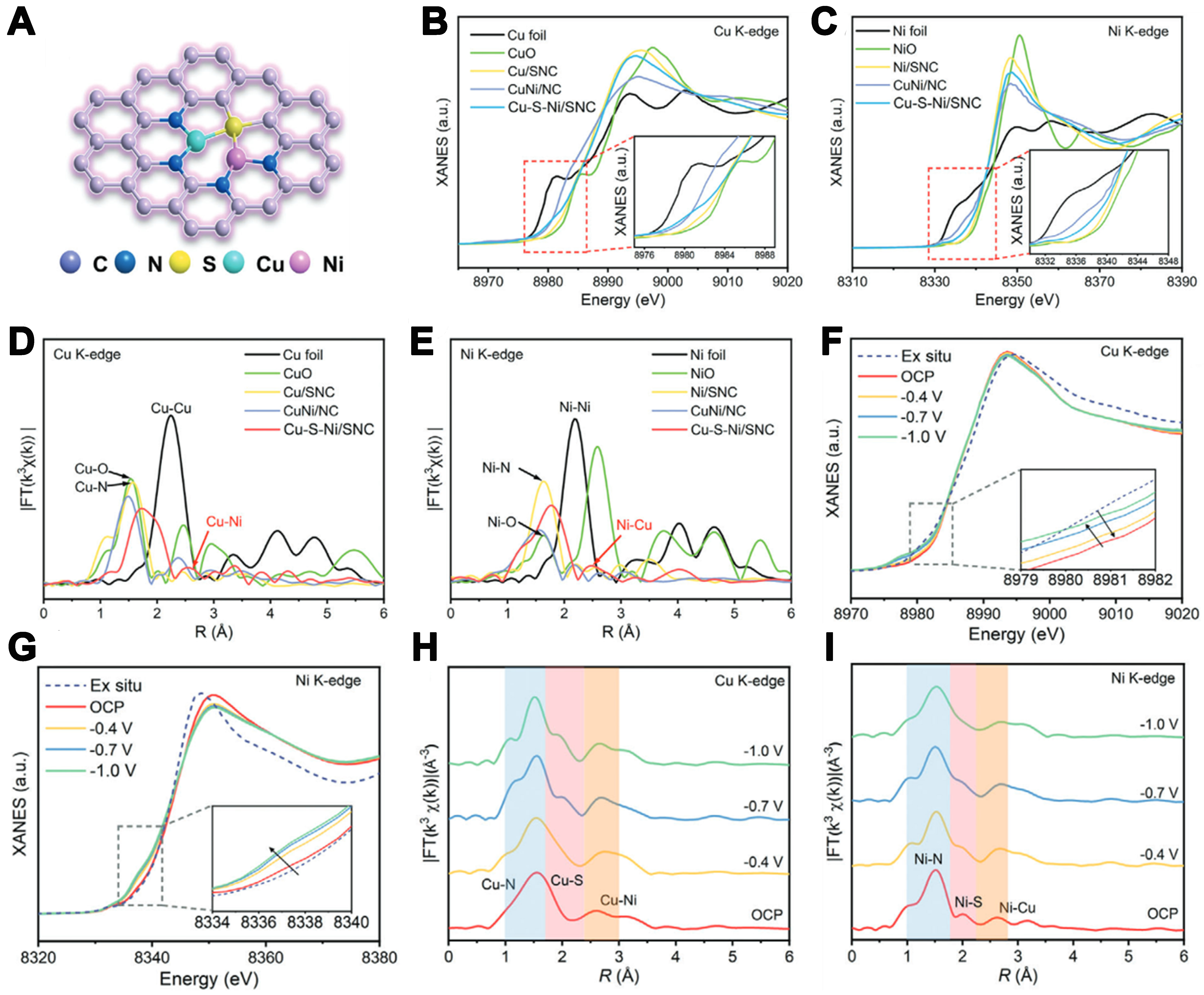
Figure 12. (A) Atomic structure model of Cu-S-Ni/SNC; (B) The Cu K edge XANES spectra of Cu-S-Ni/SNC; (C) The Ni K edge XANES spectra of Cu-S-Ni/SNC; (D) The Cu K edge FT-EXAFS spectra of Cu-S-Ni/SNC and the references; (E) The Ni K edge FT-EXAFS spectra of Cu-S-Ni/SNC and the references; (F) The Cu K-edge XANES spectra of Cu-S-Ni/SNC at various potentials during CO2RR; (G) The Ni K-edge XANES spectra of Cu-S-Ni/SNC at various potentials during CO2RR; (H) The Cu K-edge FT-EXAFS at open circuit, -0.4,
EXAFS spectra elucidate local atomic coordination in Cu–S–Ni/SNC. Cu K-edge data [Figure 12D] feature a main Cu–N peak at 1.65 Å, a Cu–S shoulder at 1.85 Å, and a minor Cu–Ni signal at 2.62 Å - longer than the 2.47 Å direct Cu–Ni bond. Ni K-edge spectra [Figure 12E] show analogous peaks at 1.72 Å (Ni-N) and
To elucidate the true active sites and their dynamic evolution, the authors employed in situ XAS spectroscopy to monitor the catalytic behavior of Cu-S-Ni/SNC [Figure 12F-I]. Upon exposure to CO2-saturated KHCO3, the Cu K-edge XANES shifts +1.05 eV, indicating CO2 adsorption–induced charge transfer and oxidation of Cu sites, while the Ni K-edge shifts -0.48 eV, suggesting Ni acts as an electron acceptor to modulate Cu’s oxidation state. Under applied potential, both Cu and Ni pre-edge peaks shift to lower binding energies, indicating progressive reduction; in Cu K-edge FT-EXAFS, the Cu–N bond contracts from 1.62 to 1.55 Å and Cu-S from 1.85 to 1.78 Å, with analogous shifts observed in the Ni EXAFS. From these observations, the authors conclude that Cu atoms serve as the principal active sites for CO2 reduction, and that S-derived electrons, coupled into the Cu/Ni 3d orbitals, synergistically enhance Cu’s adsorption and catalytic performance.
ToF-SIMS
ToF-SIMS is an advanced analytical technique that bombards a sample with high-energy ions to sputter secondary ions, which are then separated based on their mass-to-charge ratios. ToF-SIMS, as a static SIMS technique, allows for re-analysis of the sample surface without causing destruction. The mass spectra obtained not only encompass atomic ions but also include molecular ions unique to the original surface, providing richer molecular information compared to XPS and boasting higher resolution[109].
Koshy et al. synthesized Ni-N-doped carbon materials, which are designed as electrocatalysts for CO2 reduction[110]. The critical information regarding the presence and distribution of NiNxCy fragments gathered through the ToF-SIMS provided definitive evidence for the existence of isolated, nitrogen-coordinated single nickel atom sites. Specifically, within a Ni–N–C material derived from pyrolyzed polyacrylonitrile (NiPACN), a clear peak corresponding to the
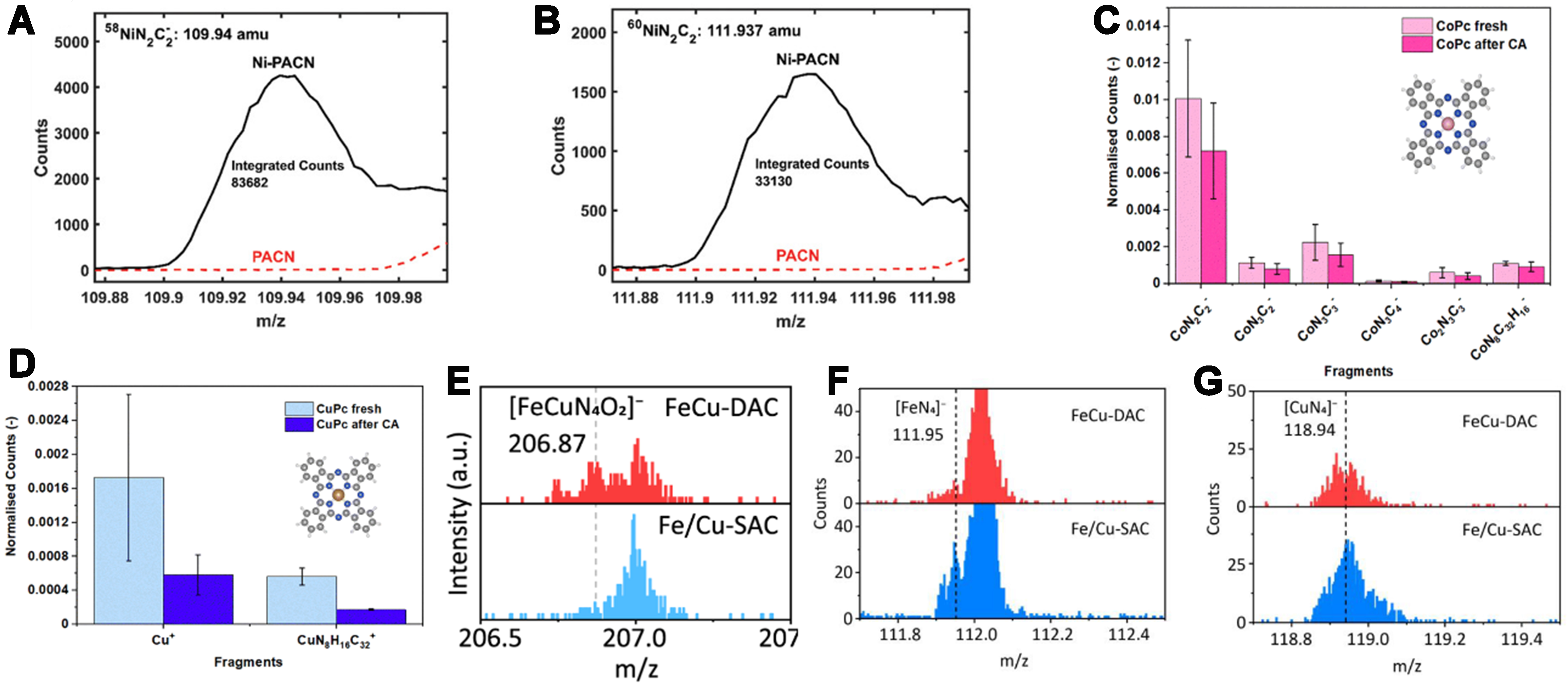
Figure 13. Comparison of ToF-SIMS intensity of NiPACN-3.5 wt% and metal-free PACN: (A) at m/z = 109.94 (58NiN2C2 weight); (B) at m/z = 111.94 (60NiN2C2 weight)[110]. Copyright 2020, Wiley-VCH; ToF-SIMS positive spectrum results of (C) Co Pc and (D) Cu Pc before and after constant potential measurements[111]. Copyright 2023, The Royal Society of Chemistry; The ToF-SIMS spectra of FeCu-DAC and Fe/Cu-SAC. Ionized fragments of (E) [FeCuN4O2]-, (F) [FeN4]-, and (G) [CuN4]- are compared for FeCu-DAC and Fe/Cu-SAC[78]. Copyright 2023, American Chemical Society. ToF-SIMS: Time-of-flight secondary ion mass spectrometry; PACN: DAC: dual-atom catalyst; SAC: single-atom catalyst.
The study by Mukadam et al. also employed ToF-SIMS as a key analytical technique, in which ToF-SIMS was used to investigate the surface composition and stability of phthalocyanine-based single-atom Co/CuPc catalysts during the electrochemical reduction of furfural[111]. By analyzing the changes in the total ion counts of Co Pc and Cu Pc before and after the constant potential testing, it was found that the normalized count of Co Pc decreased by 16% [Figure 13C], indicating the stability of Co Pc towards furfural reduction. In contrast, for Cu Pc, the normalized counts of Cu+ and Cu Pc+ decreased by 67% and 69%, respectively [Figure 13D], suggesting that Cu Pc was removed. Therefore, Cu Pc exhibits less stability towards furfural reduction compared to Co Pc. Zhang et al. employed ToF-SIMS to probe FeCu-DAC by analyzing ion-beam–ejected fragments[78]. A reference Fe/Cu-SAC was prepared via ZIF-8 pyrolysis with co-impregnated Fe and Cu nitrates. The diatomic [FeCuN4O2]- fragment exhibits markedly higher intensity in FeCu-DAC than in Fe/Cu-SAC [Figure 13E], whereas the mononuclear [FeN4]- and [CuN4]- fragments display the opposite trend [Figure 13F and G]. This unambiguously demonstrates that FeCu-DAC preserves a
CONCLUSION AND OUTLOOK
SACs with asymmetric coordination structures have emerged as a frontier in energy-related electrocatalysis owing to their tunable electronic structures and distorted coordination geometries. Unlike conventional M-N4 motifs, asymmetric SACs introduce heteroatom ligands, adjacent vacancies or multi-metal centers to break local symmetry, thereby modulating the d-band center, optimizing intermediate adsorption modes, and enabling in-situ structural reconfiguration during reaction. This often translates into markedly enhanced activities for ORR, OER, CO2RR, and beyond.
Synthesis of asymmetric SACs predominantly employs spatial-confinement strategies. MOFs, defect-rich carbon, or organic ligands serve as templates to prearrange metal precursors into predefined asymmetric sites, which are then fixed by high-temperature pyrolysis. In MOF-based routes, for instance, bimetallic complexes are assembled via electrostatic adsorption, macrocyclic-ligand mediation, or pre-coordination, encapsulated within the MOF matrix, and converted into atomically dispersed dual-atom or multi-atom centers upon carbonization. While monometallic or isolated bimetallic constructs are now well established, fabrication of heteronuclear multi-metal asymmetric sites remains challenging, necessitating precise precursor design to minimize random aggregation.
Asymmetric coordination imposes electronic perturbations that shift metal d-band centers more negatively relative to symmetric analogs, weakening undesired overbinding of oxygenated intermediates and reshaping the free-energy landscape. In multi-metal DACs, synergistic interactions between neighboring metal sites enable tandem catalysis: one site activates an intermediate while the other facilitates its conversion or desorption, thereby accelerating turnover and improving selectivity. Dynamic adaptation of active-site geometry - driven by in situ bond reorganization - further breaks linear scaling relations, unlocking higher intrinsic activities and customized product distributions.
Atomically precise SACs demand equally precise analytical tools. Aberration-corrected STEM coupled with EELS and XAS remains the workhorse for confirming SA dispersion, coordination numbers, and oxidation states. However, distinguishing subtle differences in geometric configuration, heteroatom identity, and mixed-metal composition in multi-metal SACs exceeds the capability of current techniques. Emerging methods - three-dimensional TEM/STEM tomography, in-situ scanning tunneling microscopy (STM), temporal analysis of products (TAP), and operando spectroscopies - are poised to reveal spatial heterogeneities, dynamic structural evolution, and reaction-specific active-site transformations with unprecedented resolution.
Although asymmetrically coordinated SACs have made considerable progress, they still face many challenges. (1) Precise synthesis of catalysts: High-temperature pyrolysis inherently introduces randomness, especially for heteronuclear multi-metal sites. Novel low-temperature or stepwise assembly protocols, guided by molecular-level precursors, are required to achieve uniform site construction; (2) Diversity and Scalability of catalysts: Extending beyond noble and late-transition metals to main-group and rare-earth elements could enrich the repertoire of asymmetric motifs. Moreover, scalable synthesis at gram to kilogram scale must be developed to meet industrial demands; (3) Stability of the catalyst: Unbalanced coordination numbers and heteroatom substitution can destabilize SACs under harsh ORR/OER conditions, leading to metal migration, aggregation, or carbon support corrosion. Engineering sinter-resistant supports and corrosion-resistant ligands is critical to preserve active-site integrity over prolonged cycles; (4) Selective Pathway Control: Although asymmetric sites tune adsorption energies, ensuring a single desired reaction pathway (e.g., four-electron ORR vs. two-electron H2O2 generation; CO2 - CO vs. formate vs. hydrocarbons) remains a grand challenge. Rational design - combining DFT predictions with operando validation - is needed to suppress side reactions and enhance selectivity; (5) Mechanistic insights into catalysts: Current kinetic models often oversimplify cooperative multi-metal interactions and ignore dynamic site restructuring. Integrating in situ operando characterization with advanced theoretical frameworks will be indispensable for understanding reaction mechanisms and guiding targeted catalyst optimization.
DECLARATIONS
Authors’ contributions
Conceived the project: Wan, J.; Wang, D.; Yu, R.
Wrote and edited the manuscript: Xia, T.; Wang, X.; Wan, J.; Qi, J.; Wang, D.; Yu, R.
All authors reviewed and approved the final version.
Availability of data and materials
Not applicable.
Financial support and sponsorship
This work was supported by the National Key Research and Development Program of China (No. 2024YFA1509400), National Natural Science Foundation of China (Nos. 22293043, 51932001, 51872024), and IPE Project for Frontier Basic Research, China (No. QYJC-2023-08). This work was carried out using the facilities at Huairou Interdisciplinary Research Center for Engineering Mesoscience, CAS-IPE.
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2025.
REFERENCES
1. Wei, Y.; Chen, K.; Kang, J.; Chen, W.; Wang, X.; Zhang, X. Policy and management of carbon peaking and carbon neutrality: a literature review. Engineering 2022, 14, 52-63.
2. Deng, D.; Wu, J.; Feng, Q.; et al. Highly reversible zinc-air batteries at -40 °C enabled by anion-mediated biomimetic fat. Adv. Funct. Mater. 2024, 34, 2308762.
3. Zheng, H.; Deng, D.; Zheng, X.; et al. Highly reversible Zn-air batteries enabled by tuned valence electron and steric hindrance on atomic Fe-N4-C sites. Nano. Lett. 2024, 24, 4672-81.
4. Yang, Q.; Jiang, Y.; Zhuo, H.; Mitchell, E. M.; Yu, Q. Recent progress of metal single-atom catalysts for energy applications. Nano. Energy. 2023, 111, 108404.
5. Lin, Z.; Huang, H.; Cheng, L.; et al. Tuning the p-orbital electron structure of s-block metal Ca enables a high-performance electrocatalyst for oxygen reduction. Adv. Mater. 2021, 33, e2107103.
6. Li, R.; Rao, P.; Wu, D.; et al. Understanding the bifunctional trends of Fe-based binary single-atom catalysts. Adv. Sci. 2023, 10, e2301566.
7. Zhao, Z. H.; Huang, J. R.; Liao, P. Q.; Chen, X. M. Highly efficient electroreduction of CO2 to ethanol via asymmetric C-C coupling by a metal-organic framework with heterodimetal dual sites. J. Am. Chem. Soc. 2023, 145, 26783-90.
8. Jiang, Y.; Huang, L.; Chen, C.; Zheng, Y.; Qiao, S. Catalyst–electrolyte interface engineering propels progress in acidic CO2 electroreduction. Energy. Environ. Sci. 2025, 18, 2025-49.
9. Liu, T.; Xu, T.; Li, T.; Jing, Y. Selective CO2 reduction over γ-graphyne supported single-atom catalysts: crucial role of strain regulation. J. Am. Chem. Soc. 2024, 146, 24133-40.
10. Sun, B.; Li, Z.; Xiao, D.; et al. Unveiling pH-dependent adsorption strength of *CO2- intermediate over high-density Sn single atom catalyst for acidic CO2-to-HCOOH electroreduction. Angew. Chem. Int. Ed. Engl. 2024, 63, e202318874.
11. Wang, Y.; Li, Q.; Wang, M.; et al. Pumping electrons from oxygen-bridged cobalt for low-charging-voltage Zn-air batteries. Nano. Lett. 2024, 24, 13653-61.
12. Bai, Y.; Deng, D.; Wang, J.; et al. Inhibited passivation by bioinspired cell membrane Zn interface for Zn-air batteries with extended temperature adaptability. Adv. Mater. 2024, 36, e2411404.
13. Wang, Y.; Wu, J.; Tang, S.; et al. Synergistic Fe-Se atom pairs as bifunctional oxygen electrocatalysts boost low-temperature rechargeable Zn-air battery. Angew. Chem. Int. Ed. Engl. 2023, 62, e202219191.
14. Li, Y.; Li, Y.; Sun, H.; et al. Current status and perspectives of dual-atom catalysts towards sustainable energy utilization. Nanomicro. Lett. 2024, 16, 139.
15. Wang, Y.; Su, H.; He, Y.; et al. Advanced electrocatalysts with single-metal-atom active sites. Chem. Rev. 2020, 120, 12217-314.
16. Li, Z.; Li, B.; Li, Q. Single-atom nano-islands (SANIs): a robust atomic-nano system for versatile heterogeneous catalysis applications. Adv. Mater. 2023, 35, e2211103.
17. Lyu, F.; Zeng, S.; Jia, Z.; et al. Two-dimensional mineral hydrogel-derived single atoms-anchored heterostructures for ultrastable hydrogen evolution. Nat. Commun. 2022, 13, 6249.
18. Liu, H.; Liu, C.; Zong, X.; Wang, Y.; Hu, Z.; Zhang, Z. Role of the support effects in single-atom catalysts. Chem. Asian. J. 2023, 18, e202201161.
19. Han, L.; Cheng, H.; Liu, W.; et al. A single-atom library for guided monometallic and concentration-complex multimetallic designs. Nat. Mater. 2022, 21, 681-8.
20. Jiang, Z.; Liu, X.; Liu, X. Z.; et al. Interfacial assembly of binary atomic metal-Nx sites for high-performance energy devices. Nat. Commun. 2023, 14, 1822.
21. Shen, R.; Hao, L.; Ng, Y. H.; et al. Heterogeneous N-coordinated single-atom photocatalysts and electrocatalysts. Chin. J. Catal. 2022, 43, 2453-83.
22. Wu, H.; Xu, X.; Wu, J.; et al. Atomic engineering modulates oxygen reduction of hollow carbon matrix confined single metal-nitrogen sites for zinc-air batteries. Small 2023, 19, e2301327.
23. Tian, H.; Song, A.; Zhang, P.; et al. High durability of Fe-N-C single-atom catalysts with carbon vacancies toward the oxygen reduction reaction in alkaline media. Adv. Mater. 2023, 35, e2210714.
24. Zhang, S.; Ao, X.; Huang, J.; et al. Isolated single-atom Ni-N5 catalytic site in hollow porous carbon capsules for efficient lithium-sulfur batteries. Nano. Lett. 2021, 21, 9691-8.
25. Li, Y.; Li, J.; Huang, J.; et al. Boosting electroreduction kinetics of nitrogen to ammonia via tuning electron distribution of single-atomic iron sites. Angew. Chem. Int. Ed. Engl. 2021, 60, 9078-85.
26. Lyu, L.; Hu, X.; Lee, S.; et al. Oxygen reduction kinetics of Fe-N-C single atom catalysts boosted by pyridinic N vacancy for temperature-adaptive Zn-air batteries. J. Am. Chem. Soc. 2024, 146, 4803-13.
27. Lu, X.; Li, Y.; Yang, P.; et al. Atomically dispersed Fe-N-C catalyst with densely exposed Fe-N4 active sites for enhanced oxygen reduction reaction. Chem. Eng. J. 2024, 485, 149529.
28. Yin, S.; Li, Y.; Yang, J.; et al. Unveiling low temperature assembly of dense Fe-N4 active sites via hydrogenation in advanced oxygen reduction catalysts. Angew. Chem. Int. Ed. Engl. 2024, 63, e202404766.
29. Yu, Y.; Wang, Y.; Yang, F.; et al. Meso/microporous single-atom catalysts featuring curved Fe-N4 sites boost the oxygen reduction reaction activity. Angew. Chem. Int. Ed. Engl. 2025, 64, e202415691.
30. Qin, Y.; Ou, Z.; Guo, C.; et al. Phosphor-doping modulates the d-band center of Fe atoms in Fe-N4 catalytic sites to boost the activity of oxygen reduction. Appl. Catal. B. Environ. Energy. 2025, 360, 124553.
31. Luo, X.; Wei, X.; Wang, H.; et al. Secondary-atom-doping enables robust Fe-N-C single-atom catalysts with enhanced oxygen reduction reaction. Nanomicro. Lett. 2020, 12, 163.
32. Liu, D.; Barbar, A.; Najam, T.; et al. Single noble metal atoms doped 2D materials for catalysis. Appl. Catal. B. Environ. 2021, 297, 120389.
33. Fan, M.; Cui, J.; Wu, J.; Vajtai, R.; Sun, D.; Ajayan, P. M. Improving the catalytic activity of carbon-supported single atom catalysts by polynary metal or heteroatom doping. Small 2020, 16, e1906782.
34. Chai, Y.; Dai, H.; Duan, X.; et al. Elucidation of the mechanistic origin of spin-state-dependent P-doped Fe single-atom catalysts for the oxidation of organic pollutants through peroxymonosulfate activation. Appl. Catal. B. Environ. 2024, 341, 123289.
35. Li, Y.; Wei, Z.; Sun, Z.; Zhai, H.; Li, S.; Chen, W. Sulfur modified carbon-based single-atom catalysts for electrocatalytic reactions. Small 2024, 20, e2401900.
36. Li, Z.; Wu, R.; Xiao, S.; et al. Axial chlorine coordinated iron-nitrogen-carbon single-atom catalysts for efficient electrochemical CO2 reduction. Chem. Eng. J. 2022, 430, 132882.
37. Xu, J.; Zhang, S.; Liu, H.; et al. Breaking local charge symmetry of iron single atoms for efficient electrocatalytic nitrate reduction to ammonia. Angew. Chem. Int. Ed. Engl. 2023, 62, e202308044.
38. Li, Y.; Hu, J.; Zou, Y.; et al. Catalytic activity enhancement by P and S co-doping of a single-atom Fe catalyst for peroxymonosulfate-based oxidation. Chem. Eng. J. 2023, 453, 139890.
39. Tang, F.; Zhang, G.; Wang, L.; Huang, J.; Liu, Y. Unsymmetrically N, S-coordinated single-atom cobalt with electron redistribution for catalytic hydrogenation of quinolines. J. Catal. 2022, 414, 101-8.
40. Sun, T.; Wu, Q.; Che, R.; et al. Alloyed Co–Mo nitride as high-performance electrocatalyst for oxygen reduction in acidic medium. ACS. Catal. 2015, 5, 1857-62.
41. Chen, C.; Chai, J.; Sun, M.; et al. An asymmetrically coordinated ZnCoFe hetero-trimetallic atom catalyst enhances the electrocatalytic oxygen reaction. Energy. Environ. Sci. 2024, 17, 2298-308.
42. Huang, S.; Lin, F.; Wang, S.; et al. Asymmetric microenvironment tailoring strategies of atomically dispersed dual-site catalysts for oxygen reduction and CO2 reduction reactions. Adv. Mater. 2024, 36, e2407974.
43. Cai, L.; Liu, Y.; Gao, Y.; et al. Atomically asymmetrical Ir-O-Co sites enable efficient chloride-mediated ethylene electrooxidation in neutral seawater. Angew. Chem. Int. Ed. Engl. 2025, 64, e202417092.
44. Zhan, G.; Hu, L.; Li, H.; et al. Highly selective urea electrooxidation coupled with efficient hydrogen evolution. Nat. Commun. 2024, 15, 5918.
45. Yang, X.; Song, W.; Liao, K.; et al. Cohesive energy discrepancy drives the fabrication of multimetallic atomically dispersed materials for hydrogen evolution reaction. Nat. Commun. 2024, 15, 8216.
46. Wang, Y.; Yin, H.; Dong, F.; et al. N-coordinated Cu-Ni dual-single-atom catalyst for highly selective electrocatalytic reduction of nitrate to ammonia. Small 2023, 19, e2207695.
47. Zhou, Y.; Yang, W.; Utetiwabo, W.; et al. Revealing of active sites and catalytic mechanism in N-coordinated Fe, Ni dual-doped carbon with superior acidic oxygen reduction than single-atom catalyst. J. Phys. Chem. Lett. 2020, 11, 1404-10.
48. Yu, D.; Ma, Y.; Hu, F.; et al. Dual-sites coordination engineering of single atom catalysts for flexible metal–air batteries. Adv. Energy. Mater. 2021, 11, 2101242.
49. Zhu, Z.; Yin, H.; Wang, Y.; et al. Coexisting single-atomic Fe and Ni sites on hierarchically ordered porous carbon as a highly efficient ORR electrocatalyst. Adv. Mater. 2020, 32, e2004670.
50. Li, R.; Wang, D. Superiority of dual-atom catalysts in electrocatalysis: one step further than single-atom catalysts. Adv. Energy. Mater. 2022, 12, 2103564.
51. Woldu, A. R.; Yohannes, A. G.; Huang, Z.; et al. Experimental and theoretical insights into single atoms, dual atoms, and sub-nanocluster catalysts for electrochemical CO2 reduction (CO2RR) to high-value products. Adv. Mater. 2024, 36, e2414169.
52. Zheng, X.; Liu, Y.; Yan, Y.; Li, X.; Yao, Y. Modulation effect in adjacent dual metal single atom catalysts for electrochemical nitrogen reduction reaction. Chin. Chem. Lett. 2022, 33, 1455-8.
53. Chen, C.; Sun, M.; Zhang, F.; et al. Adjacent Fe site boosts electrocatalytic oxygen evolution at Co site in single-atom-catalyst through a dual-metal-site design. Energy. Environ. Sci. 2023, 16, 1685-96.
54. Wan, J.; Zhao, Z.; Shang, H.; et al. In situ phosphatizing of triphenylphosphine encapsulated within metal-organic frameworks to design atomic Co1-P1N3 interfacial structure for promoting catalytic performance. J. Am. Chem. Soc. 2020, 142, 8431-9.
55. Li, Y.; Sun, H.; Ren, L.; et al. Asymmetric coordination regulating D-orbital spin-electron filling in single-atom iron catalyst for efficient oxygen reduction. Angew. Chem. Int. Ed. Engl. 2024, 63, e202405334.
56. Li, X.; Yang, X.; Liu, L.; et al. Chemical vapor deposition for N/S-doped single Fe site catalysts for the oxygen reduction in direct methanol fuel cells. ACS. Catal. 2021, 11, 7450-9.
57. Qu, Q.; Mao, Y.; Ji, S.; et al. Engineering the Lewis acidity of Fe single-atom sites via atomic-level tuning of spatial coordination configuration for enhanced oxygen reduction. J. Am. Chem. Soc. 2025, 147, 6914-24.
58. Ren, S.; Wang, Y.; Shi, L.; et al. Transforming plastics to single atom catalysts for peroxymonosulfate activation: axial chloride coordination intensified electron transfer pathway. Adv. Mater. 2025, 37, e2415339.
59. Yan, L.; Wang, C.; Wang, Y.; et al. Optimizing the binding of the *OOH intermediate via axially coordinated Co-N5 motif for efficient electrocatalytic H2O2 production. Appl. Catal. B. Environ. 2023, 338, 123078.
60. Liu, J.; Gong, Z.; Allen, C.; et al. Edge-hosted Fe-N3 sites on a multiscale porous carbon framework combining high intrinsic activity with efficient mass transport for oxygen reduction. Chem. Catal. 2021, 1, 1291-307.
61. Qin, Y.; Ou, Z.; Xu, C.; et al. Highly accessible single Mn-N3 sites-enriched porous graphene structure via a confined thermal-erosion strategy for catalysis of oxygen reduction. Chem. Eng. J. 2022, 440, 135850.
62. Zhang, T.; Han, X.; Liu, H.; et al. Quasi-double-star nickel and iron active sites for high-efficiency carbon dioxide electroreduction. Energy. Environ. Sci. 2021, 14, 4847-57.
63. Han, A.; Wang, X.; Tang, K.; et al. An adjacent atomic platinum site enables single-atom iron with high oxygen reduction reaction performance. Angew. Chem. Int. Ed. Engl. 2021, 60, 19262-71.
64. Zhao, L.; Cai, Q.; Mao, B.; et al. A universal approach to dual-metal-atom catalytic sites confined in carbon dots for various target reactions. Proc. Natl. Acad. Sci. U. S. A. 2023, 120, e2308828120.
65. Li, Z.; Ji, S.; Wang, C.; et al. Geometric and electronic engineering of atomically dispersed copper-cobalt diatomic sites for synergistic promotion of bifunctional oxygen electrocatalysis in zinc-air batteries. Adv. Mater. 2023, 35, e2300905.
66. Wu, J. X.; Chen, W. X.; He, C. T.; et al. Atomically dispersed dual-metal sites showing unique reactivity and dynamism for electrocatalysis. Nanomicro. Lett. 2023, 15, 120.
67. Zhang, Q.; Liu, D.; Zhang, Y.; et al. Insight into coupled Ni-Co dual-metal atom catalysts for efficient synergistic electrochemical CO2 reduction. J. Energy. Chem. 2023, 87, 509-17.
68. Wang, X.; Zhang, N.; Guo, S.; et al. p-d Orbital hybridization induced by asymmetrical FeSn dual atom sites promotes the oxygen reduction reaction. J. Am. Chem. Soc. 2024, 146, 21357-66.
69. Zhu, J.; Xiao, M.; Ren, D.; et al. Quasi-covalently coupled Ni-Cu atomic pair for synergistic electroreduction of CO2. J. Am. Chem. Soc. 2022, 144, 9661-71.
70. Pan, F.; Jin, T.; Yang, W.; et al. Theory-guided design of atomic Fe–Ni dual sites in N,P-co-doped C for boosting oxygen evolution reaction. Chem. Catal. 2021, 1, 734-45.
71. Sun, Z.; Luo, X.; Shang, H.; Wang, Z.; Zhang, L.; Chen, W. Atomic printing strategy achieves precise anchoring of dual-copper atoms on C2N structure for efficient CO2 reduction to ethylene. Angew. Chem. Int. Ed. Engl. 2024, 63, e202405778.
72. Zhang, L.; Zhang, N.; Shang, H.; et al. High-density asymmetric iron dual-atom sites for efficient and stable electrochemical water oxidation. Nat. Commun. 2024, 15, 9440.
73. Zhang, T.; Jiang, J.; Sun, W.; et al. Spatial configuration of Fe-Co dual-sites boosting catalytic intermediates coupling toward oxygen evolution reaction. Proc. Natl. Acad. Sci. U. S. A. 2024, 121, e2317247121.
74. Zhao, S.; Liu, M.; Qu, Z.; et al. Cascade synthesis of Fe-N2-Fe dual-atom catalysts for superior oxygen catalysis. Angew. Chem. Int. Ed. Engl. 2024, 63, e202408914.
75. Sun, Z.; Li, C.; Wei, Z.; et al. Sulfur-bridged asymmetric CuNi bimetallic atom sites for CO2 reduction with high efficiency. Adv. Mater. 2024, 36, e2404665.
76. Li, R.; Zhang, Z.; Liang, X.; et al. Polystyrene waste thermochemical hydrogenation to ethylbenzene by a N-bridged Co, Ni dual-atom catalyst. J. Am. Chem. Soc. 2023, 145, 16218-27.
77. Wang, B.; Yang, X.; Xie, C.; et al. A general metal ion recognition strategy to mediate dual-atomic-site catalysts. J. Am. Chem. Soc. 2024, 146, 24945-55.
78. Zhang, Y. X.; Zhang, S.; Huang, H.; et al. General synthesis of a diatomic catalyst library via a macrocyclic precursor-mediated approach. J. Am. Chem. Soc. 2023, 145, 4819-27.
79. Zhao, Y.; Chen, H. C.; Ma, X.; et al. Vacancy defects inductive effect of asymmetrically coordinated single-atom Fe-N3 S1 active sites for robust electrocatalytic oxygen reduction with high turnover frequency and mass activity. Adv. Mater. 2024, 36, e2308243.
80. Guan, G.; Liu, Y.; Li, F.; et al. Atomic cobalt metal centers with asymmetric N/B-coordination for promoting oxygen reduction reaction. Adv. Funct. Mater. 2024, 34, 2408111.
81. Yin, L.; Zhang, S.; Sun, M.; Wang, S.; Huang, B.; Du, Y. Heteroatom-driven coordination fields altering single cerium atom sites for efficient oxygen reduction reaction. Adv. Mater. 2023, 35, e2302485.
82. Shao, X.; Gan, R.; Rao, Y.; et al. Main group SnN4O single sites with optimized charge distribution for boosting the oxygen reduction reaction. ACS. Nano. 2024, 18, 14742-53.
83. Lin, X.; Zhang, X.; Liu, D.; et al. Asymmetric atomic tin catalysts with tailored p-orbital electron structure for ultra-efficient oxygen reduction. Adv. Energy. Mater. 2024, 14, 2303740.
84. Huang, M.; Deng, B.; Zhao, X.; et al. Template-sacrificing synthesis of well-defined asymmetrically coordinated single-atom catalysts for highly efficient CO2 electrocatalytic reduction. ACS. Nano. 2022, 16, 2110-9.
85. Jin, Z.; Jiao, D.; Dong, Y.; et al. Boosting electrocatalytic carbon dioxide reduction via self-relaxation of asymmetric coordination in Fe-based single atom catalyst. Angew. Chem. Int. Ed. Engl. 2024, 63, e202318246.
86. Wang, Q.; Dai, M.; Li, H.; et al. Asymmetric coordination induces electron localization at Ca sites for robust CO2 electroreduction to CO. Adv. Mater. 2023, 35, e2300695.
87. Liu, K.; Sun, Z.; Chen, W.; Lang, X.; Gao, X.; Chen, P. Ultra-fast pulsed discharge preparation of coordinatively unsaturated asymmetric copper single-atom catalysts for CO2 reduction. Adv. Funct. Mater. 2024, 34, 2312589.
88. Zhou, S.; Wei, W.; Cai, X.; et al. Customizing highly asymmetrical coordination microenvironment into P-block metal single-atom sites to boost electrocatalytic CO2 reduction. Adv. Funct. Mater. 2024, 34, 2311422.
89. Li, J.; Chen, Y.; Yao, B.; et al. Cascade dual sites modulate local CO coverage and hydrogen-binding strength to boost CO2 electroreduction to ethylene. J. Am. Chem. Soc. 2024, 146, 5693-701.
90. Li, F.; Qin, H.; Zhang, H.; et al. Another role of CO-formation catalyst in acidic tandem CO2 electroreduction: local pH modulator. Joule 2024, 8, 1772-89.
91. Chen, J.; Wang, D.; Yang, X.; et al. Accelerated transfer and spillover of carbon monoxide through tandem catalysis for kinetics-boosted ethylene electrosynthesis. Angew. Chem. Int. Ed. Engl. 2023, 62, e202215406.
92. Li, Y.; Luo, X.; Wei, Z.; et al. Precisely constructing charge-asymmetric dual-atom Fe sites supported on hollow porous carbon spheres for efficient oxygen reduction. Energy. Environ. Sci. 2024, 17, 4646-57.
93. He, N.; Sun, Y.; Chen, X.; Wang, J.; Liang, G.; Mo, F. Design of S, N-codoped Co–Fe dual-atom sites for efficient alkaline oxygen reduction. J. Mater. Chem. A. 2024, 12, 10101-9.
94. Li, L.; Zhu, J.; Kong, F.; et al. Tailoring atomic strain environment for high-performance acidic oxygen reduction by Fe-Ru dual atoms communicative effect. Matter 2024, 7, 1517-32.
95. Li, M.; Han, G.; Tian, F.; et al. Spin-polarized PdCu-Fe3O4 in-plane heterostructures with tandem catalytic mechanism for oxygen reduction catalysis. Adv. Mater. 2024, 36, e2412004.
96. Chen, C.; Sun, Z.; Qin, G.; et al. Asymmetrically coordinated Cu dual-atom-sites enables selective CO2 electroreduction to ethanol. Adv. Mater. 2024, 36, e2409797.
97. Xie, Y.; Chen, X.; Sun, K.; et al. Direct oxygen-oxygen cleavage through optimizing interatomic distances in dual single-atom electrocatalysts for efficient oxygen reduction reaction. Angew. Chem. Int. Ed. Engl. 2023, 62, e202301833.
98. Gao, Z.; Li, A.; Ma, D.; Zhou, W. Electron energy loss spectroscopy for single atom catalysis. Top. Catal. 2022, 65, 1609-19.
99. Qi, H.; Yang, J.; Liu, F.; et al. Highly selective and robust single-atom catalyst Ru1/NC for reductive amination of aldehydes/ketones. Nat. Commun. 2021, 12, 3295.
100. Qian, S.; Xu, F.; Fan, Y.; et al. Tailoring coordination environments of single-atom electrocatalysts for hydrogen evolution by topological heteroatom transfer. Nat. Commun. 2024, 15, 2774.
101. Roccapriore, K. M.; Torsi, R.; Robinson, J.; Kalinin, S.; Ziatdinov, M. Dynamic STEM-EELS for single-atom and defect measurement during electron beam transformations. Sci. Adv. 2024, 10, eadn5899.
102. Yang, J.; Liu, W.; Xu, M.; et al. Dynamic behavior of single-atom catalysts in electrocatalysis: identification of Cu-N3 as an active site for the oxygen reduction reaction. J. Am. Chem. Soc. 2021, 143, 14530-9.
103. Yang, Y.; Wang, Y.; Xiong, Y.; et al. In situ X-ray absorption spectroscopy of a synergistic Co-Mn oxide catalyst for the oxygen reduction reaction. J. Am. Chem. Soc. 2019, 141, 1463-6.
104. Gorlin, Y.; Lassalle-Kaiser, B.; Benck, J. D.; et al. In situ X-ray absorption spectroscopy investigation of a bifunctional manganese oxide catalyst with high activity for electrochemical water oxidation and oxygen reduction. J. Am. Chem. Soc. 2013, 135, 8525-34.
105. Erickson, E. M.; Thorum, M. S.; Vasić, R.; et al. In situ electrochemical X-ray absorption spectroscopy of oxygen reduction electrocatalysis with high oxygen flux. J. Am. Chem. Soc. 2012, 134, 197-200.
106. Zhu, Y.; Kuo, T.; Li, Y.; et al. Emerging dynamic structure of electrocatalysts unveiled by in situ X-ray diffraction/absorption spectroscopy. Energy. Environ. Sci. 2021, 14, 1928-58.
107. Xu, Y. N.; Mei, B.; Xu, Q.; et al. In situ/operando synchrotron radiation analytical techniques for CO2/CO reduction reaction: from atomic scales to mesoscales. Angew. Chem. Int. Ed. Engl. 2024, 63, e202404213.
108. Pei, J.; Shang, H.; Mao, J.; et al. A replacement strategy for regulating local environment of single-atom Co-SxN4-x catalysts to facilitate CO2 electroreduction. Nat. Commun. 2024, 15, 416.
109. Weng, L. Advances in the surface characterization of heterogeneous catalysts using ToF-SIMS. Appl. Catal. A. Gen. 2014, 474, 203-10.
110. Koshy, D. M.; Landers, A. T.; Cullen, D. A.; et al. Direct characterization of atomically dispersed catalysts: nitrogen-coordinated Ni sites in carbon-based materials for CO2 electroreduction. Adv. Energy. Mater. 2020, 10, 2001836.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Issue
Copyright

Data & Comments
Data
0













Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].