


Endothelial dysfunction in atherosclerosis: from classical pathways to emerging mechanisms
 0
0  ,
,
Abstract
Endothelial dysfunction is a pivotal factor in the pathogenesis of atherosclerosis, driving plaque formation, inflammation, and thrombosis. This review synthesizes classical mechanisms and emerging perspectives on endothelial dysfunction, emphasizing its role in atherogenesis. Traditional contributors, including lifestyle factors, lipid dysregulation, shear stress, and nitric oxide (NO) deficiency, are discussed alongside novel insights from single-cell RNA sequencing, metabolomics, and intercellular communication. scRNA-seq has unveiled endothelial cell (EC) heterogeneity and endothelial-to-mesenchymal transition (EndMT) as critical contributors to plaque instability. Metabolites such as trimethylamine N-oxide (TMAO) and homocysteine derivatives exacerbate endothelial injury, while gut microbiome interactions further modulate disease progression. Exosome-mediated crosstalk between ECs, immune cells, and vascular smooth muscle cells (VSMCs) highlights new pathways in vascular inflammation and remodeling. Current pharmacotherapies, such as lipid-lowering and anti-inflammatory drugs, improve endothelial function, and emerging strategies like nanotechnology and exosome-based therapies show promise as well. Integrating classical and novel approaches could enhance our understanding of endothelial biology and lead to targeted therapies, addressing atherosclerosis-related diseases.
Keywords
INTRODUCTION
Atherosclerosis is a prevalent chronic inflammatory condition of arterial walls, significantly contributing to global cardiovascular morbidity and mortality[1]. It impedes blood circulation, potentially leading to thrombus formation, which may result in heart attacks, ischemic strokes, or peripheral artery disease (PAD)[2,3]. The prevalence of coronary artery disease among U.S. adults over 20 years old is 7.1%, with myocardial infarction at 3.2%[4]. These conditions persist as primary global health threats. Atherosclerosis, initially influenced by lipoproteins, incites plaque development via intimal inflammation, necrosis, fibrosis, and calcification[3]. Over time, such plaques can induce arterial stenosis, leading to ischemic conditions, particularly in coronary, carotid, or lower limb arteries. Additionally, plaques may precipitate acute thrombosis, often due to rupture in thin-cap fibroatheromas with substantial necrotic cores, or thrombus formation from plaque erosion in pathological intimal thickening or fibroatheromas[3,5].
Endothelial dysfunction plays a critical role in the pathogenesis of atherosclerosis[6], and its role extends to related cardiovascular conditions, including myocardial ischemia-reperfusion injury and impaired microvascular flow[7,8]. Atherosclerosis originates from the subendothelial region[1]. The association between endothelial injury and atherosclerosis was initially suggested by Ross and Glomset in 1976. They introduced the “response-to-injury” hypothesis, positing that endothelial injury initiates atherogenesis[9]. This foundational theory continues to inform subsequent research[1]. By the mid-1980s, endothelial dysfunction characterized by impaired vasodilation became linked to atherosclerosis[10-12]. A significant breakthrough was the identification of endothelium-derived nitric oxide (NO), establishing the endothelium as a paracrine regulator of vasodilation and thromboprotection[13].
The endothelium functions as an organ interacting with blood flow and pressure, subject to varying flow patterns throughout the cardiac cycle. A defining characteristic of atherosclerosis is its preferential plaque formation at sites of altered hemodynamics, such as arterial branch points, rather than a uniform distribution[14]. This focal vulnerability correlates with endothelial responses to mechanical forces. The endothelium also regulates vascular homeostasis, affecting processes including vasodilation, inflammation, and coagulation. Endothelial dysfunction is characterized by impaired vascular tone and homeostasis, often indicated by reduced NO production, increased oxidative stress, and elevated inflammatory responses[15]. Compromised endothelial integrity results in lipoprotein retention, leukocyte adhesion, and smooth muscle cell proliferation, all contributing to plaque formation[16]. Endothelial inflammation promotes thrombosis, which is a key factor in adverse cardiovascular outcomes, while thrombosis, in turn, exacerbates endothelial inflammation[17]. Furthermore, clinical studies have established endothelial vasodilator dysfunction as a critical marker of atherosclerotic coronary artery disease, corroborated by animal models[18-20]. Moreover, endothelial dysfunction exacerbates myocardial ischemia-reperfusion injury by impairing microvascular perfusion and amplifying oxidative and inflammatory damage[7,21].
In addition to traditional mechanisms, recent studies utilizing cutting-edge techniques have uncovered new insights, marking a new era in the study of endothelial dysfunction in atherosclerosis. Single-cell RNA sequencing (scRNA-seq) has revealed important details about the cellular transitions of endothelial cells and their distinct roles in atherogenesis[22]. Metabolites, once considered terminal byproducts, have been found to play crucial roles in regulating cell functions, with many gut microbes contributing to this process[23]. Moreover, intercellular signaling - beyond intracellular changes - has been shown to play a critical role, leading to numerous novel findings[24]. These emerging perspectives open new avenues for understanding mechanisms and potential therapies.
In this review, we summarize both classical mechanisms and recent discoveries, with a focus on novel perspectives such as cellular heterogeneity, the roles of metabolites [Figure 1], and intercellular communication [Figure 2]. This review provides a comprehensive overview of endothelial dysfunction in atherosclerosis and offers insights into future research directions.
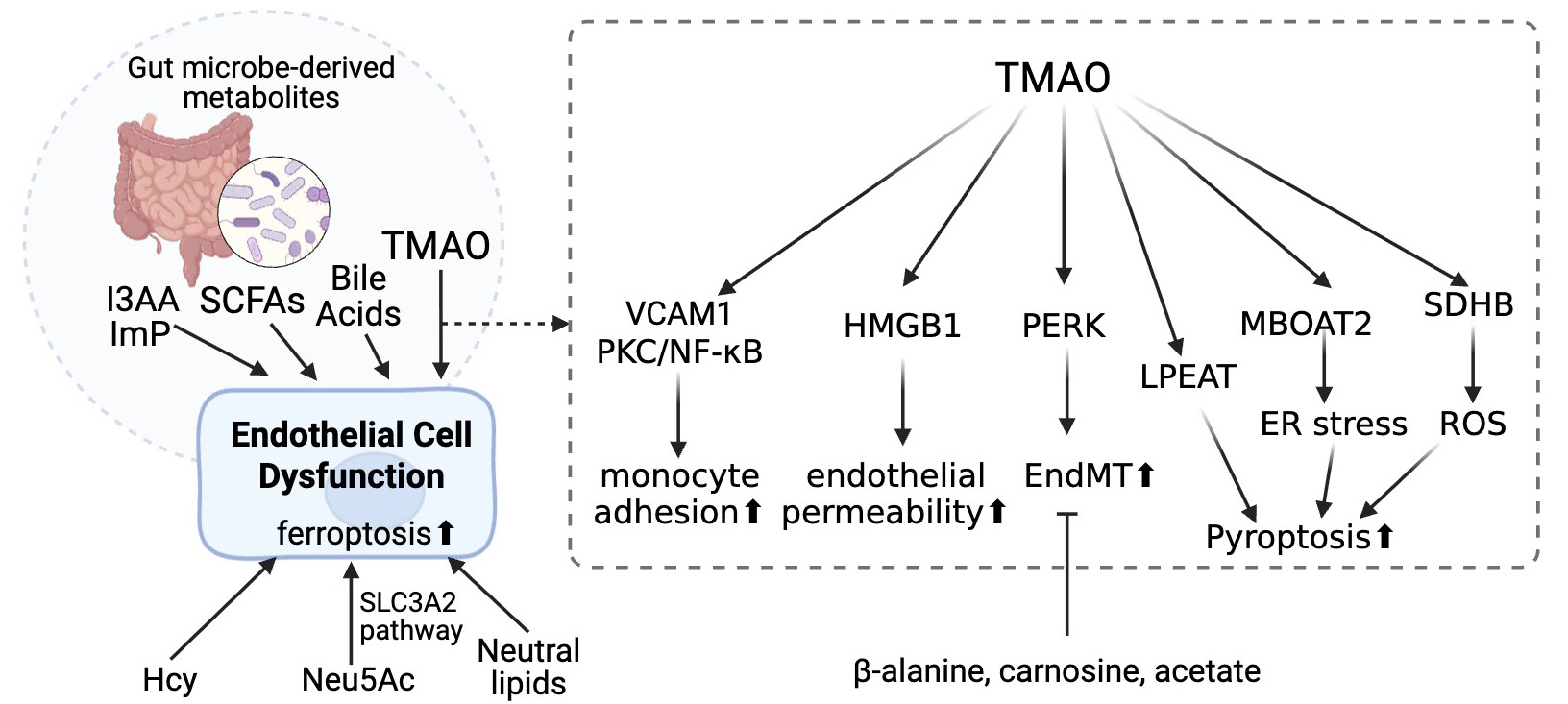
Figure 1. Gut microbiota-derived metabolites contribute to endothelial cell dysfunction through multiple pathways. Several metabolites produced by gut microbiota, including indole-3-acetic acid (I3AA), indolepropionic acid (Imp), short-chain fatty acids (SCFAs), bile acids, and trimethylamine N-oxide (TMAO), influence vascular endothelial function. These metabolites can induce endothelial cell dysfunction by promoting ferroptosis and altering cellular homeostasis. TMAO, in particular, activates various signaling pathways involving vascular cell adhesion molecule 1 (VCAM1), protein kinase C (PKC), nuclear factor kappa B (NF-κB), high mobility group box 1 (HMGB1), protein kinase RNA-like endoplasmic reticulum kinase (PERK), lysophosphatidylethanolamine acyltransferase (LPEAT), membrane-bound O-acyltransferase domain-containing 2 (MBOAT2), and succinate dehydrogenase complex iron sulfur subunit B (SDHB). These pathways contribute to increased monocyte adhesion, enhanced endothelial permeability, endothelial-to-mesenchymal transition (EndMT), endoplasmic reticulum (ER) stress, reactive oxygen species (ROS) production, and pyroptosis. Additionally, circulating factors such as homocysteine (Hcy), N-acetylneuraminic acid (Neu5Ac), and neutral lipids exacerbate endothelial dysfunction, partly through the solute carrier family 3 member 2 (SLC3A2) transport pathway. Conversely, protective metabolites including β-alanine, carnosine, and acetate may attenuate TMAO-induced endothelial injury. Created using https://BioRender.com.
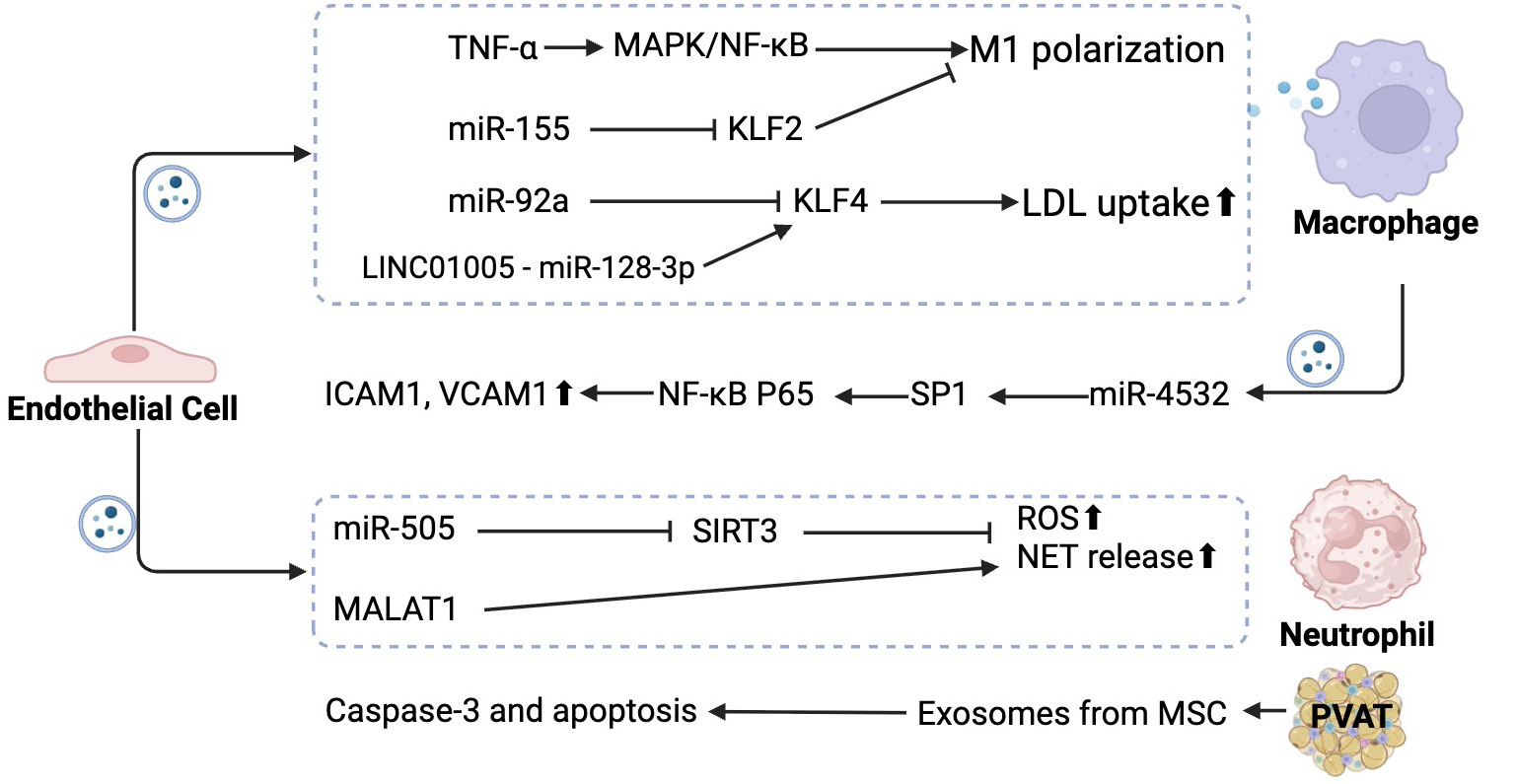
Figure 2. Endothelial-derived and incoming exosomes mediate vascular inflammation and immune cell activation in atherosclerosis. Exosomes released by endothelial cells influence the function of multiple immune and vascular cells. Endothelial cells secrete exosomes that activate the mitogen-activated protein kinase/nuclear factor kappa B (MAPK/NF-κB) pathway, promoting proinflammatory M1 polarization in macrophages. Exosomal miR-155 inhibits Kruppel-like factor 2 (KLF2), and miR-92a suppresses Kruppel-like factor 4 (KLF4), enhancing low-density lipoprotein (LDL) uptake. The long non-coding RNA LINC01005 acts as a sponge for miR-128-3p, indirectly increasing KLF4 expression and contributing to atherosclerosis progression. Exosomes derived from endothelial cells also modulate neutrophil activation. miR-505 inhibits sirtuin 3 (SIRT3), leading to elevated reactive oxygen species (ROS) and enhanced neutrophil extracellular trap (NET) release. The long non-coding RNA metastasis-associated lung adenocarcinoma transcript 1 (MALAT1), delivered by endothelial exosomes, also promotes NET formation. In the reverse direction, macrophage-derived exosomal miR-4532 targets specificity protein 1 (SP1) in endothelial cells, activating NF-κB subunit p65 and upregulating intercellular adhesion molecule 1 (ICAM1) and vascular cell adhesion molecule 1 (VCAM1), thereby promoting leukocyte adhesion. Exosomes from neutrophils and perivascular adipose tissue (PVAT) exacerbate endothelial injury. In contrast, mesenchymal stem cell (MSC)-derived exosomes protect endothelial cells by inhibiting caspase-3-mediated apoptosis, reducing oxidative stress, and restoring redox balance. Created using https://BioRender.com.
CAUSES OF ENDOTHELIAL DYSFUNCTION
Lifestyle and environmental risk factors
Lifestyle and environmental factors play a significant role in the development of endothelial dysfunction, a key contributor to atherogenesis. Modifiable risk factors, such as poor diet, physical inactivity, smoking, and exposure to environmental pollutants, can accelerate the progression of atherosclerosis. Understanding the impact of these factors is essential for developing effective strategies to prevent and treat cardiovascular disease.
Lifestyle determinants significantly contribute to the pathophysiology of endothelial dysfunction. Tobacco use represents a major modifiable risk factor that diminishes NO bioavailability while elevating the expression of adhesion molecules, consequently resulting in endothelial dysfunction and fostering a proinflammatory milieu[25]. Diets characterized by high cholesterol and saturated fat content elevate
Certain additional environmental determinants also play a significant role in the manifestation of endothelial dysfunction. Atmospheric contamination induces oxidative stress and inflammatory responses, thereby aggravating endothelial dysfunction and facilitating atherogenic transformations[32]. Moreover, psychosocial stressors[33,34] and auditory pollution[35,36] are correlated with heightened inflammatory activity and endothelial dysfunction, thereby contributing to the advancement of coronary artery disease.
While both lifestyle and environmental determinants significantly influence the advancement of endothelial dysfunction, it is crucial to evaluate the possibility of reversibility associated with this condition. Endothelial dysfunction should not be regarded as an immutable condition, as it can be ameliorated through specific interventions and modifications in lifestyle. Adjustments in lifestyle, such as the cessation of tobacco use, alterations in dietary habits, and augmentation of physical activity, have been empirically demonstrated to enhance endothelial function and mitigate cardiovascular risk[37].
Lipid products
Lipoproteins exhibit dual functions concerning the health of endothelial cells. Modified LDL and triglyceride-enriched lipoproteins induce morphological alterations in endothelial cells, manifesting as a diminished glycocalyx and heightened endoplasmic reticulum stress. These transformations, instigated by the interplay between modified lipoproteins and endothelial cells, compromise intercellular junction integrity and increase endothelial permeability[38]. Arterial injury alters these lipoproteins, facilitating their infiltration into the subendothelial compartment[1]. This accumulation incites monocyte recruitment and foam cell generation, thereby exacerbating endothelial dysfunction and the progression of atherosclerosis[39].
Variations in palmitic acid concentrations also increase oxidative stress and inflammation in endothelial cells[40]. This is evidenced by elevated levels of reactive oxygen species (ROS), primarily generated by mitochondria, Nicotinamide Adenine Dinucleotide Phosphate (NADPH) oxidase, endothelial Nitric Oxide Synthase (eNOS) uncoupling, and xanthine oxidase[41]. Additionally, the upregulation of inflammatory genes and adhesion molecules, such as IL-6, MCP-1, ICAM, and VCAM, further impairs endothelial function[40].
While lipid derivatives play a contributory role in endothelial dysfunction associated with atherosclerosis, high-density lipoprotein (HDL) exerts a protective influence by facilitating cholesterol efflux and mitigating oxidative stress under physiological conditions[42]. Conversely, under pathological circumstances, HDL becomes dysfunctional, relinquishing its protective attributes and contributing to endothelial impairment[43,44]. Dysfunctional HDL is ineffectual in inhibiting the formation of oxidized lipids, thereby exacerbating oxidative stress and endothelial dysfunction[45].
Shear stress
Endothelial cells (ECs) are perpetually subjected to hemodynamic forces resultant from blood flow, which influences their functionality across various dimensions. Unidirectional, laminar shear stress is widely regarded as a protective factor against the onset of atherosclerosis[46]. Conversely, the development of atherosclerosis is more prevalent in regions characterized by diminished wall shear stress or pulsatile disturbed shear stress, typically observed at arterial bifurcations and irregularities of the vessel wall. Such conditions facilitate the accumulation of LDL and the infiltration of inflammatory cells into the arterial wall[47]. Furthermore, these conditions inhibit the expression of the anti-inflammatory, atheroprotective transcription factor KLF2[48], which plays a crucial role in the development and progression of atherosclerotic plaques[49].
Shear stress is detected by mechanosensors, including integrins (e.g., α5-annexin A2), CD31/PECAM-1, VE-cadherin, and VEGFR2[46,50], with PECAM-1 playing a pivotal role. The tension induced by flow on PECAM-1 initiates an upstream signaling cascade that connects PECAM-1 to the vimentin cytoskeleton, thereby transmitting force from myosin. This activation triggers the engagement of Src family kinases, resulting in ligand-independent transactivation of VEGF receptors, activation of PI3K, stimulation of eNOS, and subsequent production of nitric oxide, ultimately resulting in vasodilation[51-53].
Disturbed shear stress diminishes the expression of protective genes, such as Aff3ir-ORF2, which possesses anti-inflammatory and anti-atherosclerotic properties. The lack of Aff3ir-ORF2 exacerbates inflammatory responses governed by interferon regulatory factor 5 (IRF5), fostering endothelial activation and promoting atherosclerosis[54]. Additionally, disturbed shear stress facilitates the degradation of MAPK6, an
Nonetheless, certain exceptions are noted. Disturbed flow can also activate anti-inflammatory feedback mechanisms, exemplified by the endothelial adrenomedullin-calcitonin-like receptor (CALCRL), which transmits signals via cAMP to mitigate endothelial inflammation and lesion development[57]. Although high shear stress is conventionally perceived as protective, it may also contribute to the formation of vulnerable plaques through mechanisms such as angiogenesis, thereby complicating the assertion that only low shear stress is detrimental[58].
DYSREGULATION OF ENDOTHELIAL NITRIC OXIDE IN ATHEROGENESIS
In 1980, it was demonstrated that acetylcholine-induced vasodilation necessitated an intact endothelium and was mediated by a key humoral factor, later identified as endothelium-derived relaxing factor (EDRF). Due to the similarities in both pharmacological properties[59,60] and biological effects[61,62] between EDRF and NO, EDRF was ultimately acknowledged as NO. NO is a highly unstable molecule, with a half-life of only a few seconds in oxygenated physiological environments[63]. It exerts its physiological effects by activating soluble guanylate cyclase (sGC), which elevates intracellular levels of cyclic guanosine monophosphate (cGMP) and triggers a protein kinase G (PKG) signaling cascade in vascular smooth muscle cells[64].
Synthesis of NO in endothelial cells
Endothelial cells synthesize NO through the metabolic conversion of L-arginine mediated by the eNOS[65]. The activation of eNOS transpires through a series of intricate steps. This activation is modulated by various transcriptional factors and sophisticated post-translational modification mechanisms[66,67]. In its dormant conformation, eNOS is predominantly associated with caveolin within the caveolae, which represent invaginations of the cellular membrane[68]. The binding of calcium ions (Ca2+) to calmodulin induces conformational alterations, facilitating the interaction of calmodulin with eNOS located in caveolae. This interaction results in the liberation and subsequent activation of eNOS[69]. Once situated in the cytosolic compartment, the activated eNOS catalyzes the transformation of L-arginine into NO.
The production of NO can be augmented by receptor-mediated agonists, such as acetylcholine and bradykinin, as well as non-receptor-mediated agonists like calcium ionophores, and variations in blood flow dynamics[66,70]. Moreover, hemodynamic forces exert a significant influence on the transcriptional regulation of the eNOS gene, with endothelial cells positioned in regions of stable laminar flow within arteries exhibiting enhanced NO synthesis[71,72]. Additionally, NO has the propensity to react with superoxide to yield peroxynitrite anion, which subsequently results in its inactivation.
Physiological roles of NO in the vascular system
The basal production of NO in endothelial cells is crucial for regulating vascular tone and maintaining the nonthrombogenic properties of blood vessels. Once produced, NO rapidly permeates cell membranes and functions as a potent paracrine signaling molecule.
It influences adjacent vascular smooth muscle cells (VSMC) through several mechanisms. First, it diffuses into VSMC, activating sGC to promote cGMP release, leading to VSMC relaxation[73]. Second, under hypoxic conditions, NO-induced sGC generates inosine 3’,5’-cyclic monophosphate (cIMP) instead of cGMP[74], which activates Rho-associated protein kinase (ROCK), inhibiting myosin light chain phosphatase (MLCP) and promoting VSMC contraction. Third, NO facilitates S-nitrosylation, resulting in the formation of nitrosothiols that mediate prolonged VSMC relaxation[75].
Beyond its impact on smooth muscle cells, NO also affects circulating blood platelets and leukocytes, both of which play essential roles in maintaining vascular homeostasis. Upon entering the vessel lumen, NO inhibits the activation of αIIbβ3 integrins in platelets, suppresses platelet aggregation[76,77], and obstructs the adhesion of inflammatory cells to the vessel endothelium[78]. These effects are largely mediated by sGC activation and the subsequent conversion of guanosine triphosphate (GTP) to cGMP.
Roles of NO-cGMP deficiency during the process of atherogenesis
In vivo investigations have demonstrated that mice lacking eNOS display heightened interactions between endothelial cells and leukocytes, increased platelet aggregation, and a propensity for thrombosis, as previously documented[79,80]. Additionally, Apolipoprotein E/endothelial nitric oxide synthase double-knockout mice manifest an accelerated progression of atherosclerosis[81]. Various animal models of atherosclerosis, encompassing species such as mice, rabbits, and primates, have similarly revealed that hypercholesterolemia induces significant modifications in endothelium-dependent vasodilation[19,20,82].
In human studies, coronary arteries obtained from patients undergoing cardiac transplantation, who are afflicted with coronary atherosclerosis, exhibited diminished cGMP synthesis in contrast to those without atherosclerotic conditions[19]. Furthermore, peripheral manifestations indicative of endothelial dysfunction have been recognized as independent prognosticators of atherosclerotic cardiovascular disease (ACVD) events[83]. Collectively, these observations imply that a deficiency in endothelial nitric oxide production or its bioavailability-evidenced in both human subjects and animal models-may precede the onset of clinically relevant atherosclerotic lesions.
Mechanisms of NO dysregulation
NO deficiency can occur through mechanisms such as eNOS uncoupling, abnormal phosphorylation, inhibition by endogenous methylarginines, and increased NO degradation due to ROS. These mechanisms are detailed in prior literature[84,85].
The regulation of eNOS is intricate, involving transcription, translation, protein maturation, and cofactor assembly. While eNOS activity is primarily regulated through phosphorylation, other modifications like
Furthermore, microRNAs serve as posttranscriptional regulators of nitric oxide release from endothelial cells by influencing eNOS[87]. In endothelial cells, miR-92a modulates eNOS mRNA levels by targeting Klf2[88]. Additionally, miR-221 and miR-222 expression increases during early atherosclerosis, negatively correlating with eNOS signaling[89]. Moreover, miR-195 and miR-532, potential biomarkers for thrombosis, inversely relate to eNOS expression[87].
ENDOTHELIAL CELLULAR STATE TRANSITION DURING ATHEROSCLEROSIS
With the advancement of research, several novel perspectives have emerged. While classical mechanisms involved in endothelial dysfunction, such as oxidative stress, inflammation, hemodynamic disturbances, apoptosis, and cell death, have been briefly discussed above and extensively explored in previous reviews[6,16], the focus here will be on newer mechanisms.
Endothelial-to-mesenchymal transition
Endothelial-to-mesenchymal transition (EndMT) is a major feature of atherosclerotic plaque development and was reported to drive the development and progression of atherosclerosis[90]. EndMT is marked by reduced endothelial markers (e.g., CD31, NOS3) and increased mesenchymal markers [e.g., fibroblast activation protein (FAP), alpha-actin 2 (ACTA)][91]. Lineage-tracing experiments in ApoE−/− mice on a
Various molecular pathways and factors have been identified as key regulators of EndMT. In vitro studies have demonstrated that EndMT is induced by TGF-β signaling, oxidative stress, and hypoxia[91,94]. Similar to TGF-β signaling, fibroblast growth factor (FGF) pathways are activated by inflammation and shear stress, but endothelial FGF receptor (FGFR) signaling offers protection against atherosclerosis[90]. In ApoE-/- mice, the absence of FGFR substrate 2a leads to enhanced plaque burden and neointima formation, driven by excessive EndMT and fibronectin accumulation[90]. The USF1/USP14/NLRC5 axis has been identified as a promoter of EndMT in atherosclerosis, triggering the activation of the Smad2/3 signaling cascade, which is essential for EndMT progression. Silencing USF1 can block this pathway, thus reducing EndMT and atherosclerosis development in vitro and in vivo[95]. Furthermore, a study using scRNA-seq identified ENO1, regulated by disturbed flow, as a key player in EC inflammation and EndMT in a shear flow model[96]. ENO1 knockdown in human aortic endothelial cells (HAECs) upregulated EC markers (CD31 and CDH5) while downregulating mesenchymal markers induced by disturbed flow[96].
Epigenetic regulation, including histone modification and DNA methylation, plays a crucial role in controlling the expression levels of endothelial-specific genes and upstream modulators that regulate EndMT. The molecular mechanisms reported have been comprehensively summarized in the review by Singh[97]. Recent studies demonstrated the role of epigenetics in EndMT in atherosclerotic mouse models. Histone lactylation, particularly at H3K18la, promotes EndMT and atherosclerosis. This modification is regulated by the ASF1A/P300 complex, which enhances SNAI1 transcription. Inhibiting glycolysis and ASF1A reduces H3K18la levels, decreasing EndMT and offering a potential therapeutic approach for atherosclerosis[98]. Lecce et al. found that the knockout of HDAC9 in endothelial cells prevented EndMT and maintained an endothelial-like phenotype[99]. In vivo, mice with endothelial-specific HDAC9 knockout showed reduced EndMT and smaller plaque areas[99]. In line with this, another study found that valproic acid (VPA), an HDAC inhibitor, induced TGFβ-mediated EndMT in endothelial cells[100]. Additionally, DNA methyltransferases (DNMTs) have been recognized as crucial molecular mediators of EndMT.
EC heterogeneity revealed by scRNA-seq
In addition to the well-known EndMT state of ECs, scRNA-seq has significantly advanced the understanding of EC heterogeneity in atherosclerosis by providing detailed insights into the diverse cellular phenotypes and functions within atherosclerotic lesions[22,102]. A comprehensive scRNA-seq study identified more than 78 distinct EC clusters across 11 murine tissues[103]. Kalluri et al. demonstrated that two EC subsets have specialized functions in lipoprotein handling, angiogenesis, and extracellular matrix production[104]. He et al. identified three primary EC populations in the murine aorta: CD34high ECs, THY1high ECs, and “activated ECs”[105]. While CD34high and THY1high ECs are primarily associated with endothelial proliferation and vascular development, activated ECs are enriched in pathways related to vessel dilation and blood pressure regulation[105]. Another study highlighted the functional heterogeneity of ECs in atherosclerosis, showing that SULF1+(sulfatase-1) endothelial cells contribute to plaque vulnerability and cerebrovascular events through proinflammatory activities[106].
METABOLIC SIGNALING PATHWAYS
Effects of key metabolites
Recent studies have highlighted the significant role of metabolites in influencing endothelial function and atherogenesis [Figure 1], with homocysteine metabolites being particularly impactful[107]. Elevated levels of homocysteine (Hcy) and its metabolites, such as Hcy-thiolactone and N-Hcy-protein, are strongly associated with endothelial dysfunction and cardiovascular diseases (CVD)[107]. Hcy-thiolactone modifies lysine residues on proteins, resulting in the formation of N-Hcy-proteins, which are cytotoxic, proinflammatory, and proatherogenic[108]. These metabolites trigger pro-atherogenic changes in gene expression, activate mTOR signaling, and inhibit autophagy through epigenetic mechanisms, all of which contribute to endothelial dysfunction[109,110]. Even small physiological increases in plasma total homocysteine (tHcy), induced by low-dose methionine or dietary animal protein, have been shown to impair vascular endothelial function. This suggests that diet-induced elevations in plasma tHcy may contribute to the progression of atherosclerosis by compromising vascular function[111].
Another metabolite, Neu5Ac, has been shown to promote endothelial ferroptosis via the SLC3A2 pathway, contributing to endothelial cell injury and accelerating atherosclerotic plaque progression in Apoe-/- mice[112]. Additionally, supplementation with functional metabolites such as β-alanine, carnosine, and acetyl-CoA (acetate) has been found to reverse EndMT, a key process in atherogenesis. This effect is likely mediated through the inhibition of Smad2/3 phosphorylation[101]. Furthermore, the accumulation of neutral lipids resulting from the deletion of adipose triglyceride lipase (ATGL) in the endothelium leads to impaired endothelial-dependent vascular tone and reduced nitric oxide synthesis, exacerbating endothelial dysfunction. Loss of ATGL also induces endoplasmic reticulum stress, which triggers inflammation in the endothelium and increases lesion size in atherosclerosis models[113].
Gut microbiome metabolism
A variety of metabolites derived from the gut microbiota play a crucial role in the progression of atherosclerosis, including short-chain fatty acids (SCFAs), trimethylamine N-oxide (TMAO), secondary bile acids (BAs), and other molecules[17,114-116].
TMAO induces endothelial dysfunction through multiple mechanisms[117]. Querio et al. systematically investigated how it induces endothelial dysfunction[118]. Using bovine aortic endothelial cell line (BAE-1 cells) as a model, the results showed that TMAO altered the purinergic response, which means it affected intracellular ATP-induced calcium increase, nitric oxide release, and phosphorylation of eNOS.
The expression of adhesion molecules is also regulated by TMAO. It increases monocyte adhesion by upregulating vascular cell adhesion molecule 1 (VCAM-1) and activates the protein kinase C (PKC)/nuclear factor kappa B (NF-κB) pathway, thereby promoting atherogenesis[119]. Additionally, TMAO increases endothelial permeability and dysfunction by disrupting cell junctions through the action of high-mobility group box protein 1 (HMGB1)[120]. Furthermore, TMAO induces EndMT and apoptosis through protein kinase RNA-like endoplasmic reticulum kinase (PERK) signaling, worsening plaque formation and cardiac function[121]. Finally, TMAO promotes vascular inflammation by activating mitogen-activated protein kinase (MAPK) and NF-κB pathways, enhancing inflammatory gene expression and leukocyte recruitment, which increases the risk of atherosclerosis[122].
Studies have also shown that TMAO affects pyroptosis, a form of programmed cell death associated with inflammation. This process involves the lysophosphatidylethanolamine acyltransferase (LPEAT) and mitophagy pathways, leading to excessive cell death and promoting the progression of atherosclerosis[23]. In ApoE−/− mice, TMAO accelerates atherosclerosis by triggering pyroptosis through membrane-bound
In addition to TMAO, certain SCFAs produced by specific microbiota species from indigestible carbohydrates also contribute to endothelial inflammation[17]. The mechanisms by which SCFAs are involved in atherosclerosis have been extensively reviewed[114-116]. Recent studies have focused more on the therapeutic effects of microbiome-associated SCFAs. For instance, Zhu et al. found that E. faecium NCIMB11508 positively impacted gut microbiota composition by increasing SCFA levels, which, in turn, reduced inflammation and protected the intestine[125]. Yang et al. demonstrated that dietary methionine restriction (MR) promoted SCFA production and bile acid metabolism, reduced cell adhesion molecules, and prevented foam cell formation in the aorta, thereby helping to prevent atherosclerosis[126].
Additionally, other gut microbiome-derived metabolites are involved in atherogenesis. Indole-3-acrylic acid (I3AA), a metabolite derived from microbes, plays a key role in alleviating atherosclerosis (AS) by activating the aryl hydrocarbon receptor (AhR) and inhibiting the TGF-β/Smad pathway. This may help mitigate endothelial-to-mesenchymal transitions in atherosclerotic mice[127]. In Apoe-/- mice, imidazole propionate (ImP) was found to increase atherosclerotic lesion size. Mechanistically, ImP disrupts insulin receptor signaling by inhibiting the PI3K/AKT pathway, leading to prolonged activation of the FOXO1 (forkhead box protein O1) transcription factor[128].
INTERCELLULAR COMMUNICATION AND EXOSOME-MEDIATED CROSSTALK
Endothelial-derived exosomes as key mediators of vascular crosstalk
Exosomes, which can carry small molecules, serve as mediators in communication among various cell types and play a role in endothelial dysfunction and atherosclerosis [Figure 2]. Endothelial-derived exosomes can activate macrophages, a crucial cell type in atherosclerotic plaques[129]. When endothelial cells are exposed to inflammatory cytokines like TNF-α, they release exosomes that promote a proinflammatory M1 phenotype in macrophages through the MAPK/NF-κB signaling pathway. This polarization exacerbates lipid accumulation and triggers macrophage apoptosis, contributing to plaque instability[130]. Several studies have identified specific molecules within exosomes derived from endothelial cells. MiR-155, for example, was found to be highly expressed in exosomes from both oxLDL-treated HUVECs and VSMCs, driving proinflammatory M1 polarization in THP-1 monocytes[131]. This effect occurs through the downregulation of KLF2, a repressor of miR-155, resulting in elevated levels of miR-155 in both cells and exosomes. KLF2 is known for its anti-inflammatory role in endothelial cells during atherogenesis. MiR-92a, contained in exosomes derived from endothelial cells, downregulated the expression of its target gene KLF4 in macrophages. This led to enhanced proinflammatory responses, increased LDL uptake, and impaired macrophage migration. The study further demonstrated that these harmful effects were alleviated in both miR-92a knockdown and KLF4 overexpression models. Observations in atherosclerotic Apoe-/- mice revealed an inverse correlation between intravesicular miR-92a levels in blood serum and KLF4 expression in lesions, suggesting a potential role for extracellular miR-92a in the formation of atherosclerotic lesions[132].
Exosomes released from ECs play a crucial role in regulating the phenotypic changes, proliferation, and migration of VSMCs, which are pivotal in the development of atherosclerosis[133,134]. The long non-coding RNA LINC01005, highly expressed in oxLDL-treated HUVECs, is also abundant in exosomes derived from these cells[135]. LINC01005 promotes the phenotypic shift, proliferation, and migration of VSMCs. Mechanistically, LINC01005 functions as a sponge for miR-128-3p, leading to the upregulation of its target gene KLF4. These findings indicate that LINC01005-mediated modulation of the miR-128-3p/KLF4 axis in exosomes from oxLDL-treated HUVECs reduces contractile markers while increasing the synthetic smooth muscle cell marker osteopontin (OPN), thereby contributing to the progression of atherosclerosis[135].
Exosomes derived from endothelial cells can be transferred to immune cells, where they influence immune cell function[136]. For instance, exosomes containing interleukin-6 (IL-6) can promote the differentiation of Th17 cells, leading to increased production of inflammatory cytokines and contributing to the development of atherosclerosis[137]. Metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) has also been identified as a factor that activates neutrophil extracellular traps (NETs). It is delivered to neutrophils via exosomes released by HUVECs treated with oxLDL. In atherosclerotic Apoe-/- mice, treatment with exosomes from oxLDL-stimulated HUVECs triggered an inflammatory response and promoted NET release, thereby exacerbating atherosclerosis[138]. Additionally, the upregulation of miR-505, which is encapsulated within exosomes from HUVECs, inhibits Sirtuin 3 (SIRT3) in neutrophils. This inhibition increases ROS production and further enhances NET formation by neutrophils[139].
Reverse communication: neighboring cells to endothelium
Exosomes derived from various cell types and organs, including macrophages, mesenchymal stem cells, dendritic cells, and perivascular adipose tissue (PVAT), can exacerbate endothelial dysfunction in atherosclerosis[136].
For instance, macrophage-derived exosomal miR-4532 has been shown to promote endothelial cell injury by targeting SP1 and activating NF-κB p65 signaling, leading to increased expression of adhesion molecules such as ICAM-1 and VCAM-1, which facilitate leukocyte adhesion and transmigration[140]. MicroRNAs involved in the inflammatory response of monocytes, miR-155 and miR-233, are significantly altered within these exosomes. The target of these exosomes is primarily endothelial cells, which were shown to effectively internalize these bioactive vesicles, subsequently causing an increase in the expression of NF-κB[141].
MSC-derived exosomes were internalized by oxLDL-treated ECs, where they inhibited caspase-3 activation and cell apoptosis, promoted EC proliferation, and reduced proinflammatory cytokines and oxidative stress levels. Additionally, they increased antioxidant levels (SOD and GSH-PX), offering protection against oxLDL-induced EC dysfunction[142]. Furthermore, exosomes derived from adipose-derived mesenchymal stem cells (ADMSCs) were shown to protect ECs from atherosclerosis. In a H2O2-treated HUVEC lesion model, miR-324-5p expression was decreased, and miR-324-5p inhibited the expression of Protein Phosphatase 1 Regulatory Subunit 12B (PPP1R12B), highlighting its protective role[143].
Endothelial cells can also internalize platelet-derived exosomes. One example is miR-223, which inhibits ICAM-1 expression and protects atherosclerotic ApoE-/- mice from endothelial inflammation[144].
Myoendothelial junction
Myoendothelial junctions (MEJs) are specialized structures where endothelial cells directly interface with vascular smooth muscle cells, enabling intercellular signaling that regulates vascular tone and blood pressure[145]. MEJs enable the transfer of ions and small molecules, which are essential for coordinating vasodilation and vasoconstriction[146]. Dysfunction of MEJs impairs vasodilation, increases vascular resistance, and contributes to the progression of atherosclerosis.
Endothelial cells, sensitive to hemodynamic changes, activate signaling pathways involving ion channels and NO production to respond to changes in blood flow and wall tension[147]. MEJs provide a direct signaling pathway between endothelial cells and VSMCs, particularly for transporting small molecules such as Ca2+ and IP3[148,149]. This signaling forms the “myoendothelial feedback” (MEF) loop, where endothelial NO release and Ca2+-activated K⁺ channels help inhibit excessive VSMC contraction, thereby reducing vascular tension[147]. The localization of signaling proteins within MEJs further emphasizes their critical role in vascular regulation.
Continuous mechanical strain can induce phenotypic changes and functional remodeling of VSMCs, altering the structure of the vascular wall and its ability to adapt to stimuli[150,151]. This mechanical regulation is essential for maintaining vascular function and ensuring stable blood flow.
PHARMACOTHERAPIES TARGETING ENDOTHELIAL DYSFUNCTION IN ATHEROSCLEROSIS
Accumulating evidence suggests that pharmacological agents-ranging from approved therapeutics to investigational compounds, with diverse chemical structures and mechanisms of action-can modulate endothelial function. This section provides an overview of classical pharmacological agents and summarizes some novel drugs with promising potential and related clinical trials [Tables 1 and 2].
Pharmacotherapies targeting endothelial dysfunction
| Drug class | Agent | Mechanisms influencing endothelial function |
| Lipid-lowering drugs | Statins (e.g., Rosuvastatin, Simvastatin) | ↑ NO, ↓ ROS, ↓ CRP, improved FMD |
| PCSK9 inhibitors (e.g., Evolocumab) | ↓ Oxidative stress & inflammation, improved endothelial function | |
| Antihypertensive drugs | ARBs (e.g., Losartan) | ↓ Endothelin-1, ↓ inflammation |
| ACEIs (e.g., Enalapril) | Improved FMD in multiple vascular beds | |
| Direct renin inhibitor (Aliskiren) | ↓ EPCs, no FMD improvement | |
| Calcium channel blockers (e.g., Amlodipine) | Restore endothelium-dependent vasodilation | |
| β-blockers (Nebivolol vs. Atenolol) | Nebivolol: beneficial; Atenolol: negative | |
| Anti-diabetic drugs | GLP-1 receptor agonists | ↑ NO, ↓ inflammation, improved FMD |
| SGLT-2 inhibitors | Improved endothelial markers, mechanism unclear | |
| Metformin | ↑ insulin sensitivity, ↓ ROS | |
| TZDs (e.g., Rosiglitazone) | PPAR-γ activation, anti-inflammatory | |
| Insulin analogs | Mechanisms unclear | |
| Anti-inflammatory drugs | Aspirin | Improved endothelium-dependent vasodilation |
| IL-35 | ↓ Vascular inflammation | |
| Flavonoids, antioxidant mimetics | ↓ Oxidative stress, improved vascular function (animal) | |
| Colchicine | NLRP3 inhibition, improved endothelial markers | |
| MCC950 | NLRP3 inhibition (preclinical) | |
| Resolvin D1 (RvD1) | ↓ NF-κB, ↓ adhesion molecules | |
| eNOS enhancers/antioxidants | BH4 + antioxidants | Restore eNOS coupling, ↑ NO |
| Resveratrol | ↑ eNOS via Nrf2, ↓ ROS | |
| Vitamin C & E | Inhibit NAD(P)H oxidase, ↑ FMD | |
| AVE3085 | ↑ eNOS transcription, ↓ oxidative stress | |
| N-acetylcysteine (NAC) | Antioxidant effect (preclinical) | |
| Genistein | ↑ eNOS, ↑ calmodulin-1, ↓ ROS | |
| Novel strategies | β2 receptor agonists | ↑ eNOS, ↑ NO, ↓ ROS |
| Molsidomine | Direct NO donor, no effect in angina | |
| KCa channel activators | ↑ NO, ↓ oxidative stress | |
| AMPK activators | ↓ lipid inflammation, ↑ NO | |
| Exosomal circ_0001785 | ↓ EC injury via miR-513a-5p/TGFBR3 |
Clinical studies on endothelial function and atherosclerosis
| Study (PMID) | Drug | Target | Disease | Sample size | Results |
| 21349594 | Rosuvastatin | HMG-CoA reductase | STEMI | 87 | Positive |
| 29209775 | Rosuvastatin | HMG-CoA reductase | CAD | 50 | Positive |
| 20199786 | Simvastatin | HMG-CoA reductase | PreDM | 50 | Positive |
| 37468450 | Evolocumab | PCSK9 | ACS | 46 | Positive |
| 23615219 | Aliskiren | renin | EA | 22 | No effect |
| 30683457 | Colchicine | NLRP3 | CAD | 28 | Positive in part |
| 25875387 | Molsidomine | NO | SAP | 180 | No effect |
Lipid-lowering drug
Lipid-lowering agents, especially statins, are highly effective in improving endothelial dysfunction. Their primary mechanisms involve increasing NO bioavailability and reducing oxidative stress, thereby preserving NO function and maintaining normal vasodilation[6,37]. Furthermore, statins markedly diminish the generation of ROS, thus mitigating oxidative stress that contributes to endothelial injury[152]. Additionally, statins exhibit notable anti-inflammatory properties by substantially lowering inflammatory markers such as C-reactive protein (CRP), thereby enhancing vascular stability[153].
Numerous clinical investigations have demonstrated that statin therapy markedly improves endothelial biomarkers, particularly in individuals with metabolic syndrome and dyslipidemia. Prolonged administration of statins, such as rosuvastatin, leads to sustained enhancement of endothelial function and reduced lipid peroxidation, which may promote the regression of atherosclerotic lesions. High-dose rosuvastatin treatment improves endothelium-dependent flow-mediated dilation (FMD) of the brachial artery in patients with ST-segment elevation myocardial infarction (STEMI)[154]. Additionally, aggressive lowering of low-density lipoprotein cholesterol (LDL-C) with rosuvastatin significantly improved the reactive hyperemia peripheral arterial tonometry (RH-PAT) index, suggesting that it may improve endothelial function in patients with coronary artery disease[155]. Previous studies have suggested that simvastatin may help preserve endothelial function in prediabetic individuals with normal or mildly to moderately elevated cholesterol levels[156]. Continued inquiry into the molecular underpinnings of endothelial dysfunction, alongside the multifaceted effects of lipid-lowering treatments such as statins, presents promising prospects for developing innovative therapeutic strategies to improve endothelial health[6].
PCSK9 inhibitors (PCSK9i), an emergent lipid-lowering modality, attenuate endothelial dysfunction and platelet activation in patients with acute coronary syndrome[157]. In the context of atherosclerosis, PCSK9i enhance endothelial function by abating oxidative stress and inflammation, thus counteracting vascular aging and dysfunction[158].
Antihypertensive drugs
Angiotensin II receptor blockers (ARBs) and angiotensin-converting enzyme inhibitors (ACEIs) have demonstrated positive effects on endothelial function. Drugs such as losartan can reduce levels of endothelin-1 and other serum inflammatory markers, thereby alleviating endothelial dysfunction. ACEIs have also been shown to improve endothelial function in multiple vascular regions[159]. A meta-analysis of 38 clinical trials found that, compared to other antihypertensive drugs, ACE inhibitors and ARBs are more effective in improving endothelial function, as measured by FMD, although conflicting data exist among these studies[160]. Additionally, a four-month treatment with the direct renin inhibitor aliskiren was shown to decrease circulating endothelial progenitor cells (EPCs) without affecting endothelial function[161]. Calcium channel blockers, particularly the dihydropyridine class, can also significantly reverse endothelium-dependent vasodilation impairment. In contrast, traditional β-blockers such as atenolol may have negative effects on endothelial function, whereas newer β-blockers like nebivolol show beneficial effects[162].
Anti-diabetic drugs
Various anti-diabetic drugs not only exert their effects through glycemic control but also show independent benefits in improving endothelial function. For instance, GLP-1 receptor agonists, SGLT-2 inhibitors, and thiazolidinediones (TZDs) can enhance NO production, inhibit inflammatory cytokines, and reduce oxidative stress, significantly improving endothelial function indicators such as flow-mediated dilation[163]. Metformin reduces endothelial injury by increasing insulin sensitivity and scavenging free radicals[164], while TZDs such as rosiglitazone activate the PPAR-γ pathway, exert anti-inflammatory effects, and improve vascular responsiveness[165]. These mechanisms help restore endothelial cell physiological function, potentially slow the progression of atherosclerosis, and reduce cardiovascular risk[166]. However, the mechanisms of action for certain agents, such as insulin analogs and some SGLT-2 inhibitors, remain incompletely understood, and further clinical studies are needed to verify their long-term efficacy and cardiovascular outcomes. Future research should focus on comprehensively evaluating the multifaceted vascular effects of anti-diabetic drugs to support more personalized and targeted therapeutic strategies.
Anti-inflammatory drugs
Given the central role of inflammation in endothelial dysfunction and the development and progression of atherosclerosis, anti-inflammatory drugs have garnered significant attention in recent years. Widely used anti-inflammatory agents, such as aspirin, have been shown to improve endothelium-dependent vasodilation in high-risk individuals and patients with established atherosclerosis[167]. Emerging anti-inflammatory cytokines, like interleukin-35 (IL-35), have also been identified as potential therapeutic targets, offering new avenues for managing vascular inflammation in CVD[168]. Additionally, natural compounds with both antioxidant and anti-inflammatory properties-such as flavonoids and antioxidant enzyme mimetics-have demonstrated promise in reducing oxidative stress and improving vascular function in animal models[169].
As research continues to highlight the importance of NLRP3 (NOD-like receptor protein 3) as a key driver of atherosclerosis, drugs targeting this pathway are gaining attention. According to a recent review, the NLRP3 inflammasome plays a crucial role in endothelial dysfunction, with its activation triggered by factors such as ROS, ischemia, and high glucose, leading to inflammation and endothelial injury[170]. The NLRP3 inflammasome inhibitor MCC950 has shown promising results in preclinical studies; however, its clinical utility still needs to be confirmed[171]. Colchicine, another inhibitor of the NLRP3 inflammasome, has been tested in clinical trials and shown to improve endothelial function in patients with leukocyte activation, although it had no effect on endothelial function in the overall patient population[172]. NLRP3 can be activated by oxLDL, mitochondrial ROS, and TMAO[173]. Upon activation, NLRP3 assembles the inflammasome, activates caspase-1, and promotes the release of interleukin-1β (IL-1β) and interleukin-18 (IL-18)[174,175]. These cytokines upregulate endothelial adhesion molecules and enhance leukocyte adhesion. NLRP3 inflammasome can also be regulated by gut microbiota, which warrants further investigation[176].
Endogenous agents are also becoming better understood and show considerable promise. For example, specialized pro-resolving mediators derived from DHA and EPA, such as resolvin D1 (RvD1), have demonstrated promising effects in suppressing NF-κB activation and reducing adhesion molecule expression, effectively mitigating endothelial inflammation induced by stimuli like cholesterol crystals[177,178].
eNOS enhancers and antioxidant agents
Various pharmacological agents and natural substances enhance vascular endothelial function by mitigating oxidative stress, restoring eNOS coupling, and augmenting NO bioavailability. Antioxidants contribute to this process by scavenging free radicals and supporting the availability of essential enzyme cofactors, thus preserving eNOS activity and promoting NO production[179].
Both BH4 and its combinations with antioxidants have been validated in research to enhance eNOS performance[180]. Moreover, interventions targeting eNOS regulation-such as inhibiting VE-PTP or utilizing resveratrol-loaded nanoparticles-have shown promise in augmenting NO production and minimizing oxidative stress[181]. Resveratrol enhances eNOS expression and activity via Nrf2 activation, BH4 elevation, and ROS inhibition[182]. Vitamin C facilitates the conversion of oxidized BH4 back to its active state and supports sustained NO release[180]. In healthy subjects, vitamins C and E have been demonstrated to enhance FMD by inhibiting NAD(P)H oxidase, leading to improved endothelial function[183]. The compound AVE3085 has proven effective in ameliorating hypertension and endothelial dysfunction by promoting eNOS transcription and coupling, inhibiting oxidative stress, and reducing arginase activity[184-186]. Furthermore, N-acetylcysteine (NAC) has shown promising effects on endothelial function in preclinical investigations[187]. Genistein supports endothelial integrity by enhancing eNOS activity, increasing calmodulin-1 expression, and scavenging ROS[188].
Novel potential therapeutic strategies
Beta2 (B2) receptor agonists, recently discovered drugs, show promise in treating endothelial dysfunction, particularly in cardiovascular diseases. They improve endothelial function by enhancing NO production, stimulating eNOS activity, reducing oxidative stress, preventing hypoxia/reoxygenation injury, and improving vascular permeability[189]. However, a clinical study found that add-on treatment with Molsidomine, a direct NO donor, did not improve endothelial function in patients with stable angina pectoris[190].
Enhancing the activity of endothelial Ca2+-activated K+ (KCa) channels[191] and activating AMP-activated protein kinase (AMPK)[192] are also promising strategies to restore endothelial function. These approaches aim to reduce oxidative stress, improve NO bioavailability, and counteract the proinflammatory effects of lipid products.
As summarized in the previous section on exosomes, certain exosomal components have demonstrated protective effects in atherosclerotic endothelial dysfunction. For instance, exosome-derived circ_0001785 has been shown to reduce endothelial cell injury and delay atherogenesis through the miR-513a-5p/TGFBR3 ceRNA network, highlighting a potential therapeutic strategy for atherosclerosis[193].
FUTURE DIRECTIONS
Endothelial cell heterogeneity and spatial omics
Emerging single-cell and spatial transcriptomic technologies have revealed substantial heterogeneity among ECs across different vascular beds, with distinct subpopulations associated with plaque rupture susceptibility, lipid transcytosis, and leukocyte adhesion[22,102]. Future research should focus on deepening our understanding of the functions of each EC subtype. Furthermore, integrating spatial transcriptomics and/or spatial proteomics to map EC subtypes within human atherosclerotic lesions-especially in high-risk areas like plaque shoulders and necrotic cores-will be crucial. Combining these approaches with in vivo shear stress modeling could provide insights into how oscillatory flow drives proinflammatory EC phenotypes via mechanosensitive pathways. Additionally, lineage-tracing models could offer further evidence in these studies.
Nanotechnology for targeted therapeutics
Nanoparticle-based strategies offer unprecedented precision for modulating endothelial dysfunction. A huge advantage of nanoparticle delivery is that it can target certain cell types, avoiding unwanted effects. For example, dysregulation of YAP/TAZ contributes to plaque development, making them potential therapeutic targets. However, systemic modulation of YAP/TAZ poses risks of off-target effects. To overcome this, a study developed monocyte membrane-coated nanoparticles (MoNP) for targeted delivery to activated, inflamed endothelium on plaque surfaces, effectively reducing plaque development in mice without causing significant histopathological changes in major organs[194]. Another study developed a biomimetic
Clinical translation and trials
As more mechanisms underlying endothelial dysfunction in atherogenesis are uncovered, specific therapies warrant further investigation in clinical trials to assess their potential for clinical application. For instance, microbiome-modulating interventions, such as prebiotics that promote butyrate production, may enhance endothelial barrier function. Long-term follow-up studies are crucial to determine whether short-term endothelial repair leads to a reduction in major adverse cardiovascular events (MACE).
CONCLUSION
The endothelium plays a crucial role as both a sensor of hemodynamic stress and a regulator of vascular homeostasis, making it central to the pathogenesis of atherosclerosis. While traditional approaches targeting LDL-C and blood pressure remain essential, cutting-edge strategies could further reveal the mechanisms of endothelial heterogeneity, mechanotransduction, and metabolic reprogramming. Advances in gene editing, nanomedicine, and systems biology provide new tools to develop spatially resolved, cell-type-specific therapies. However, significant gaps remain in improving endothelial function and preventing adverse events due to a limited understanding of critical mechanisms such as redox-inflammation coupling and endothelial pyroptosis. These complex pathological processes highlight the need for multi-target therapies and more precise treatments. By prioritizing translational studies that connect endothelial biology with clinical outcomes, the field can move beyond plaque stabilization and aim for true vascular repair, offering hope for reversing atherosclerosis in its early stages.
DECLARATIONS
Acknowledgments
The authors express gratitude to Dr. Deqiang Kong, Dr. Yiyun Xie, and Dr. Qijian Zhao for their valuable discussions and insights regarding the content of this work. All figures were created using https://Biorender.com.
Authors’ contributions
Conceptualized and supervised the review composition: Liu B, Wang J, Zhao H
Wrote the first draft of the manuscript and revised the manuscript: Feng Y, Li C
Created the figures and the graphical abstract: Feng Y
Created the tables: Li C
Reviewed and edited the manuscript: Chen J, Xiao X, Mao Q
All authors reviewed and approved the final manuscript.
Availability of data and materials
Not applicable.
Financial support and sponsorship
This work was supported by the National Natural Science Foundation of China (No. 82470516) and Beijing Municipal Natural Science Foundation (No. L248071).
Conflicts of interest
Zhao H is a Youth Editorial Board member of the journal Vessel Plus. Zhao H was not involved in any steps of editorial processing, notably including reviewer selection, manuscript handling, or decision making. The other authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2025.
REFERENCES
2. Hansson GK, Libby P. The immune response in atherosclerosis: a double-edged sword. Nat Rev Immunol. 2006;6:508-19.
3. Bentzon JF, Otsuka F, Virmani R, Falk E. Mechanisms of plaque formation and rupture. Circ Res. 2014;114:1852-66.
4. Martin SS, Aday AW, Allen NB, et al; American Heart Association Council on Epidemiology and Prevention Statistics Committee and Stroke Statistics Committee. 2025 heart disease and stroke statistics: a report of US and global data from the American Heart Association. Circulation. 2025;151:e41-e660.
5. Gaba P, Gersh BJ, Muller J, Narula J, Stone GW. Evolving concepts of the vulnerable atherosclerotic plaque and the vulnerable patient: implications for patient care and future research. Nat Rev Cardiol. 2023;20:181-96.
6. Xu S, Ilyas I, Little PJ, et al. Endothelial dysfunction in atherosclerotic cardiovascular diseases and beyond: from mechanism to pharmacotherapies. Pharmacol Rev. 2021;73:924-67.
7. Welt FGP, Batchelor W, Spears JR, et al. Reperfusion injury in patients with acute myocardial infarction: JACC scientific statement. J Am Coll Cardiol. 2024;83:2196-213.
8. Konijnenberg LSF, Damman P, Duncker DJ, et al. Pathophysiology and diagnosis of coronary microvascular dysfunction in ST-elevation myocardial infarction. Cardiovasc Res. 2020;116:787-805.
9. Ross R, Glomset JA. The pathogenesis of atherosclerosis (first of two parts). N Engl J Med. 1976;295:369-77.
12. Anderson TJ, Gerhard MD, Meredith IT, et al. Systemic nature of endothelial dysfunction in atherosclerosis. Am J Cardiol. 1995;75:71B-4B.
13. Furchgott RF. The discovery of endothelium-derived relaxing factor and its importance in the identification of nitric oxide. Jama. ;1996:276:1186-8.
14. Glagov S, Zarins C, Giddens DP, et al. Hemodynamics and atherosclerosis. Insights and perspectives gained from studies of human arteries. Arch Pathol Lab Med. 1988;112:1018-31.
15. Mussbacher M, Schossleitner K, Kral-Pointner JB, Salzmann M, Schrammel A, Schmid JA. More than just a monolayer: the multifaceted role of endothelial cells in the pathophysiology of atherosclerosis. Curr Atheroscler Rep. 2022;24:483-92.
16. Gimbrone MA Jr, García-Cardeña G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res. 2016;118:620-36.
17. Cimmino G, Muscoli S, De Rosa S, et al; Pathogenesis Of Atherosclerosis Working Group Of The Italian Society Of Cardiology. Evolving concepts in the pathophysiology of atherosclerosis: from endothelial dysfunction to thrombus formation through multiple shades of inflammation. J Cardiovasc Med. 2023;24:e156-67.
18. Ludmer PL, Selwyn AP, Shook TL, et al. Paradoxical vasoconstriction induced by acetylcholine in atherosclerotic coronary arteries. N Engl J Med. 1986;315:1046-51.
19. Bossaller C, Habib GB, Yamamoto H, Williams C, Wells S, Henry PD. Impaired muscarinic endothelium-dependent relaxation and cyclic guanosine 5’-monophosphate formation in atherosclerotic human coronary artery and rabbit aorta. J Clin Invest. 1987;79:170-4.
20. d’Uscio LV, Smith LA, Katusic ZS. Hypercholesterolemia impairs endothelium-dependent relaxations in common carotid arteries of apolipoprotein e-deficient mice. Stroke. 2001;32:2658-64.
21. Ghanta SN, Kattamuri LPV, Odueke A, Mehta JL. Molecular insights into ischemia-reperfusion injury in coronary artery disease: mechanisms and therapeutic implications: a comprehensive review. Antioxidants. 2025;14:213.
22. de Winther MPJ, Bäck M, Evans P, et al. Translational opportunities of single-cell biology in atherosclerosis. Eur Heart J. 2023;44:1216-30.
23. Chen Y, Yuan C, Qin W, et al. TMAO promotes vascular endothelial cell pyroptosis via the LPEAT-mitophagy pathway. Biochem Biophys Res Commun. 2024;703:149667.
24. Wehbe Z, Wehbe M, Al Khatib A, et al. Emerging understandings of the role of exosomes in atherosclerosis. J Cell Physiol. 2025;240:e31454.
25. Messner B, Bernhard D. Smoking and cardiovascular disease: mechanisms of endothelial dysfunction and early atherogenesis. Arterioscler Thromb Vasc Biol. 2014;34:509-15.
26. Mente A, de Koning L, Shannon HS, Anand SS. A systematic review of the evidence supporting a causal link between dietary factors and coronary heart disease. Arch Intern Med. 2009;169:659-69.
27. Renaud S, Lanzmann-Petithory D. Coronary heart disease: dietary links and pathogenesis. Public Health Nutr. 2001;4:459-74.
28. Shao C, Wang J, Tian J, Tang Y. Coronary artery disease: from mechanism to clinical practice. In: Wang M, editor. Coronary Artery Disease: Therapeutics and drug discovery. Singapore: Springer; 2020. pp. 1-36.
30. De Bosscher R, Dausin C, Claus P, et al. Lifelong endurance exercise and its relation with coronary atherosclerosis. Eur Heart J. 2023;44:2388-99.
31. Nishitani-Yokoyama M, Miyauchi K, Shimada K, et al. Impact of physical activity on coronary plaque volume and components in acute coronary syndrome patients after early phase ii cardiac rehabilitation. Circ J. 2018;83:101-9.
32. Tibuakuu M, Michos ED, Navas-Acien A, Jones MR. Air pollution and cardiovascular disease: a focus on vulnerable populations worldwide. Curr Epidemiol Rep. 2018;5:370-8.
33. Hinterdobler J, Schott S, Jin H, et al. Acute mental stress drives vascular inflammation and promotes plaque destabilization in mouse atherosclerosis. Eur Heart J. 2021;42:4077-88.
34. Wirtz PH, von Känel R. Psychological stress, inflammation, and coronary heart disease. Curr Cardiol Rep. 2017;19:111.
35. Chen X, Liu M, Zuo L, et al. Environmental noise exposure and health outcomes: an umbrella review of systematic reviews and meta-analysis. Eur J Public Health. 2023;33:725-31.
36. Malakar AK, Choudhury D, Halder B, Paul P, Uddin A, Chakraborty S. A review on coronary artery disease, its risk factors, and therapeutics. J Cell Physiol. 2019;234:16812-23.
37. Poredos P, Poredos AV, Gregoric I. Endothelial dysfunction and its clinical implications. Angiology. 2021;72:604-15.
38. Stancu CS, Toma L, Sima AV. Dual role of lipoproteins in endothelial cell dysfunction in atherosclerosis. Cell Tissue Res. 2012;349:433-46.
39. Gofman JW, Lindgren F. The role of lipids and lipoproteins in atherosclerosis. Science. 1950;111:166-71.
40. Rhee M, Lee J, Lee EY, Yoon KH, Lee SH. Lipid variability induces endothelial dysfunction by increasing inflammation and oxidative stress. Endocrinol Metab. 2024;39:511-20.
41. Incalza MA, D’Oria R, Natalicchio A, Perrini S, Laviola L, Giorgino F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vascul Pharmacol. 2018;100:1-19.
42. Terasaka N, Yu S, Yvan-Charvet L, et al. ABCG1 and HDL protect against endothelial dysfunction in mice fed a high-cholesterol diet. J Clin Invest. 2008;118:3701-13.
43. Besler C, Heinrich K, Rohrer L, et al. Mechanisms underlying adverse effects of HDL on eNOS-activating pathways in patients with coronary artery disease. J Clin Invest. 2011;121:2693-708.
44. Nicholls SJ, Zheng L, Hazen SL. Formation of dysfunctional high-density lipoprotein by myeloperoxidase. Trends Cardiovasc Med. 2005;15:212-9.
45. Eren E, Yilmaz N, Aydin O. Functionally defective high-density lipoprotein and paraoxonase: a couple for endothelial dysfunction in atherosclerosis. Cholesterol. 2013;2013:792090.
46. Zhang C, Zhou T, Chen Z, et al. Coupling of integrin α5 to annexin A2 by flow drives endothelial activation. Circ Res. 2020;127:1074-90.
47. Zhou M, Yu Y, Chen R, et al. Wall shear stress and its role in atherosclerosis. Front Cardiovasc Med. 2023;10:1083547.
48. Nigro P, Abe J, Berk BC. Flow shear stress and atherosclerosis: a matter of site specificity. Antioxid Redox Signal. 2011;15:1405-14.
49. Wang X, Shen Y, Shang M, Liu X, Munn LL. Endothelial mechanobiology in atherosclerosis. Cardiovasc Res. 2023;119:1656-75.
50. Baeyens N, Bandyopadhyay C, Coon BG, Yun S, Schwartz MA. Endothelial fluid shear stress sensing in vascular health and disease. J Clin Invest. 2016;126:821-8.
51. Conway DE, Breckenridge MT, Hinde E, Gratton E, Chen CS, Schwartz MA. Fluid shear stress on endothelial cells modulates mechanical tension across VE-cadherin and PECAM-1. Curr Biol. 2013;23:1024-30.
52. Tzima E, Irani-Tehrani M, Kiosses WB, et al. A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature. 2005;437:426-31.
53. Fleming I, Fisslthaler B, Dixit M, Busse R. Role of PECAM-1 in the shear-stress-induced activation of Akt and the endothelial nitric oxide synthase (eNOS) in endothelial cells. J Cell Sci. 2005;118:4103-11.
54. He S, Huang L, Chen Z, Yuan Z, Zhao Y, Zeng L, Zhu Y, He J. Disruption of the novel nested gene aff3ir mediates disturbed flow-induced atherosclerosis in mice. eLife. 2025;13:RP103413.
55. Wang F, Wang SY, Gu Y, et al. Disturbed shear stress promotes atherosclerosis through TRIM21-regulated MAPK6 degradation and consequent endothelial inflammation. Clin Transl Med. 2025;15:e70168.
56. Lv Y, Jiang Z, Zhou W, et al. Low-shear stress promotes atherosclerosis via inducing endothelial cell pyroptosis mediated by IKKε/STAT1/NLRP3 Pathway. Inflammation. 2024;47:1053-66.
57. Nakayama A, Albarrán-Juárez J, Liang G, et al. Disturbed flow-induced Gs-mediated signaling protects against endothelial inflammation and atherosclerosis. JCI Insight. 2020;5:140485.
58. Wang Y, Qiu J, Luo S, et al. High shear stress induces atherosclerotic vulnerable plaque formation through angiogenesis. Regen Biomater. 2016;3:257-67.
59. Ignarro LJ, Buga GM, Wood KS, Byrns RE, Chaudhuri G. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc Natl Acad Sci U S A. 1987;84:9265-9.
60. Ignarro LJ, Buga GM, Wei LH, Bauer PM, Wu G, del Soldato P. Role of the arginine-nitric oxide pathway in the regulation of vascular smooth muscle cell proliferation. Proc Natl Acad Sci U S A. 2001;98:4202-8.
61. Hutchinson PJ, Palmer RM, Moncada S. Comparative pharmacology of EDRF and nitric oxide on vascular strips. Eur J Pharmacol. 1987;141:445-51.
62. Radomski MW, Palmer RM, Moncada S. Comparative pharmacology of endothelium-derived relaxing factor, nitric oxide and prostacyclin in platelets. Br J Pharmacol. 1987;92:181-7.
63. Griffith TM, Edwards DH, Lewis MJ, Newby AC, Henderson AH. The nature of endothelium-derived vascular relaxant factor. Nature. 1984;308:645-7.
64. Rapoport RM, Murad F. Agonist-induced endothelium-dependent relaxation in rat thoracic aorta may be mediated through cGMP. Circ Res. 1983;52:352-7.
65. Sessa WC, Harrison JK, Barber CM, et al. Molecular cloning and expression of a cDNA encoding endothelial cell nitric oxide synthase. J Biol Chem. 1992;267:15274-6.
67. Dudzinski DM, Michel T. Life history of eNOS: partners and pathways. Cardiovasc Res. 2007;75:247-60.
68. Bucci M, Gratton JP, Rudic RD, et al. In vivo delivery of the caveolin-1 scaffolding domain inhibits nitric oxide synthesis and reduces inflammation. Nat Med. 2000;6:1362-7.
69. Sandoo A, van Zanten JJ, Metsios GS, Carroll D, Kitas GD. The endothelium and its role in regulating vascular tone. Open Cardiovasc Med J. 2010;4:302-12.
70. Kuchan MJ, Frangos JA. Role of calcium and calmodulin in flow-induced nitric oxide production in endothelial cells. Am J Physiol. 1994;266:C628-36.
71. Gimbrone MA Jr, García-Cardeña G. Vascular endothelium, hemodynamics, and the pathobiology of atherosclerosis. Cardiovasc Pathol. 2013;22:9-15.
72. Chiu JJ, Chien S. Effects of disturbed flow on vascular endothelium: pathophysiological basis and clinical perspectives. Physiol Rev. 2011;91:327-87.
73. Lee MR, Li L, Kitazawa T. Cyclic GMP causes Ca2+ desensitization in vascular smooth muscle by activating the myosin light chain phosphatase. J Biol Chem. 1997;272:5063-8.
74. Gao Y, Chen Z, Leung SW, Vanhoutte PM. Hypoxic vasospasm mediated by cIMP: when soluble guanylyl cyclase turns bad. J Cardiovasc Pharmacol. 2015;65:545-8.
75. Kraehling JR, Sessa WC. Contemporary Approaches to Modulating the Nitric Oxide-cGMP Pathway in Cardiovascular Disease. Circ Res. 2017;120:1174-82.
76. Procter NE, Chong CR, Sverdlov AL, Chan WP, Chirkov YY, Horowitz JD. Aging of platelet nitric oxide signaling: pathogenesis, clinical implications, and therapeutics. Semin Thromb Hemost. 2014;40:660-8.
77. Radomski MW, Palmer RM, Moncada S. Modulation of platelet aggregation by an L-arginine-nitric oxide pathway. Trends Pharmacol Sci. 1991;12:87-8.
78. Bath PM. The effect of nitric oxide-donating vasodilators on monocyte chemotaxis and intracellular cGMP concentrations in vitro. Eur J Clin Pharmacol. 1993;45:53-8.
79. Kuhlencordt PJ, Rosel E, Gerszten RE, et al. Role of endothelial nitric oxide synthase in endothelial activation: insights from eNOS knockout endothelial cells. Am J Physiol Cell Physiol. 2004;286:C1195-202.
80. Huang PL. Lessons learned from nitric oxide synthase knockout animals. Semin Perinatol. 2000;24:87-90.
81. Kuhlencordt PJ, Gyurko R, Han F, et al. Accelerated atherosclerosis, aortic aneurysm formation, and ischemic heart disease in apolipoprotein E/endothelial nitric oxide synthase double-knockout mice. Circulation. 2001;104:448-54.
82. Freiman PC, Mitchell GG, Heistad DD, Armstrong ML, Harrison DG. Atherosclerosis impairs endothelium-dependent vascular relaxation to acetylcholine and thrombin in primates. Circ Res. 1986;58:783-9.
83. Davignon J, Ganz P. Role of endothelial dysfunction in atherosclerosis. Circulation. 2004;109:III27-32.
84. Förstermann U, Sessa WC. Nitric oxide synthases: regulation and function. Eur Heart J. 2012;33:829-37.
85. Garcia V, Sessa WC. Endothelial NOS: perspective and recent developments. Br J Pharmacol. 2019;176:189-96.
86. Aulak KS, Barnes JW, Tian L, et al. Specific O-GlcNAc modification at Ser-615 modulates eNOS function. Redox Biol. 2020;36:101625.
87. Qin JZ, Wang SJ, Xia C. microRNAs regulate nitric oxide release from endothelial cells by targeting NOS3. J Thromb Thrombolysis. 2018;46:275-82.
88. Bonauer A, Carmona G, Iwasaki M, et al. MicroRNA-92a controls angiogenesis and functional recovery of ischemic tissues in mice. Science. 2009;324:1710-3.
89. Chistiakov DA, Sobenin IA, Orekhov AN, Bobryshev YV. Human miR-221/222 in physiological and atherosclerotic vascular remodeling. Biomed Res Int. 2015;2015:354517.
90. Chen PY, Qin L, Baeyens N, et al. Endothelial-to-mesenchymal transition drives atherosclerosis progression. J Clin Invest. 2015;125:4514-28.
91. Evrard SM, Lecce L, Michelis KC, et al. Endothelial to mesenchymal transition is common in atherosclerotic lesions and is associated with plaque instability. Nat Commun. 2016;7:11853.
92. Zhao G, Lu H, Liu Y, et al. Single-cell transcriptomics reveals endothelial plasticity during diabetic atherogenesis. Front Cell Dev Biol. 2021;9:689469.
93. Huang Q, Gan Y, Yu Z, Wu H, Zhong Z. Endothelial to mesenchymal transition: an insight in atherosclerosis. Front Cardiovasc Med. 2021;8:734550.
94. Chen PY, Qin L, Li G, et al. Endothelial TGF-β signalling drives vascular inflammation and atherosclerosis. Nat Metab. 2019;1:912-26.
95. Zhang Z, Guo Q, Ma C, et al. USF1 transcriptionally activates USP14 to drive atherosclerosis by promoting EndMT through NLRC5/Smad2/3 axis. Mol Med. 2024;30:32.
96. Chen LJ, Li JY, Nguyen P, et al. Single-cell RNA sequencing unveils unique transcriptomic signatures of endothelial cells and role of ENO1 in response to disturbed flow. Proc Natl Acad Sci U S A. 2024;121:e2318904121.
97. Singh A, Bhatt KS, Nguyen HC, Frisbee JC, Singh KK. Endothelial-to-mesenchymal transition in cardiovascular pathophysiology. Int J Mol Sci. 2024;25:6180.
98. Dong M, Zhang Y, Chen M, et al. ASF1A-dependent P300-mediated histone H3 lysine 18 lactylation promotes atherosclerosis by regulating EndMT. Acta Pharm Sin B. 2024;14:3027-48.
99. Lecce L, Xu Y, V’Gangula B, et al. Histone deacetylase 9 promotes endothelial-mesenchymal transition and an unfavorable atherosclerotic plaque phenotype. J Clin Invest. 2021:131.
100. Murugavel S, Bugyei-Twum A, Matkar PN, et al. Valproic acid induces endothelial-to-mesenchymal transition-like phenotypic switching. Front Pharmacol. 2018;9:737.
101. Zhao J, Zhao C, Yang F, et al. DNMT1 mediates the disturbed flow-induced endothelial to mesenchymal transition through disrupting β-alanine and carnosine homeostasis. Theranostics. 2023;13:4392-411.
102. Wakabayashi T, Naito H. Cellular heterogeneity and stem cells of vascular endothelial cells in blood vessel formation and homeostasis: Insights from single-cell RNA sequencing. Front Cell Dev Biol. 2023;11:1146399.
103. Kalucka J, de Rooij LPMH, Goveia J, et al. Single-cell transcriptome atlas of murine endothelial cells. Cell. 2020;180:764-779.e20.
104. Kalluri AS, Vellarikkal SK, Edelman ER, et al. Single-Cell analysis of the normal mouse aorta reveals functionally distinct endothelial cell populations. Circulation. 2019;140:147-63.
105. He D, Mao A, Zheng CB, et al. Aortic heterogeneity across segments and under high fat/salt/glucose conditions at the single-cell level. Natl Sci Rev. 2020;7:881-96.
106. Tan J, Liang Y, Yang Z, et al. Single-cell transcriptomics reveals crucial cell subsets and functional heterogeneity associated with carotid atherosclerosis and cerebrovascular events. Arterioscler Thromb Vasc Biol. 2023;43:2312-32.
107. Jakubowski H, Witucki Ł. Homocysteine metabolites, endothelial dysfunction, and cardiovascular disease. Int J Mol Sci. 2025;26:746.
108. Jakubowski H. Homocysteine modification in protein structure/function and human disease. Physiol Rev. 2019;99:555-604.
109. Gurda D, Handschuh L, Kotkowiak W, Jakubowski H. Homocysteine thiolactone and N-homocysteinylated protein induce pro-atherogenic changes in gene expression in human vascular endothelial cells. Amino Acids. 2015;47:1319-39.
110. Witucki Ł, Jakubowski H. Homocysteine metabolites impair the PHF8/H4K20me1/mTOR/autophagy pathway by upregulating the expression of histone demethylase PHF8-targeting microRNAs in human vascular endothelial cells and mice. FASEB J. 2024;38:e70072.
111. Jakubowski H, Zhang L, Bardeguez A, Aviv A. Homocysteine thiolactone and protein homocysteinylation in human endothelial cells: implications for atherosclerosis. Circ Res. 2000;87:45-51.
112. Xiang P, Chen Q, Chen L, et al. Metabolite Neu5Ac triggers SLC3A2 degradation promoting vascular endothelial ferroptosis and aggravates atherosclerosis progression in ApoE-/- mice. Theranostics. 2023;13:4993-5016.
113. Boutagy NE, Gamez-Mendez A, Fowler JW, et al. Dynamic metabolism of endothelial triglycerides protects against atherosclerosis in mice. J Clin Invest. 2024:134.
114. Brown JM, Hazen SL. Microbial modulation of cardiovascular disease. Nat Rev Microbiol. 2018;16:171-81.
115. Qian B, Zhang K, Li Y, Sun K. Update on gut microbiota in cardiovascular diseases. Front Cell Infect Microbiol. 2022;12:1059349.
116. Luqman A, Hassan A, Ullah M, et al. Role of the intestinal microbiome and its therapeutic intervention in cardiovascular disorder. Front Immunol. 2024;15:1321395.
117. Querio G, Antoniotti S, Geddo F, Levi R, Gallo MP. Modulation of endothelial function by TMAO, a gut microbiota-derived metabolite. Int J Mol Sci. 2023;24:5806.
118. Querio G, Antoniotti S, Geddo F, Levi R, Gallo MP. Trimethylamine N-oxide (TMAO) impairs purinergic induced intracellular calcium increase and nitric oxide release in endothelial Cells. Int J Mol Sci. 2022;23:3982.
119. Ma G, Pan B, Chen Y, et al. Trimethylamine N-oxide in atherogenesis: impairing endothelial self-repair capacity and enhancing monocyte adhesion. Biosci Rep. 2017;37:BSR20160244.
120. Singh GB, Zhang Y, Boini KM, Koka S. High mobility group box 1 mediates TMAO-induced endothelial dysfunction. Int J Mol Sci. 2019;20:3570.
121. Bingyu W, Jun Q, Bingyang L, Xi Y, Jianqing Z, Jiangfang L. Trimethylamine N-oxide promotes PERK-mediated endothelial-mesenchymal transition and apoptosis thereby aggravates atherosclerosis. Int Immunopharmacol. 2024;142:113209.
122. Seldin MM, Meng Y, Qi H, et al. Trimethylamine N-oxide promotes vascular inflammation through signaling of mitogen-activated protein kinase and nuclear factor-κB. J Am Heart Assoc. 2016;5:e002767.
123. Yu B, Yuan C, Chen J, et al. TMAO induces pyroptosis of vascular endothelial cells and atherosclerosis in ApoE-/- mice via MBOAT2-mediated endoplasmic reticulum stress. Biochim Biophys Acta Mol Cell Biol Lipids. 2024;1869:159559.
124. Wu P, Chen J, Chen J, et al. Trimethylamine N-oxide promotes ApoE-/- mice atherosclerosis by inducing vascular endothelial cell pyroptosis via the SDHB/ROS pathway. J Cell Physiol. 2020;235:6582-91.
125. Zhu Y, Yin C, Wang Y. Probiotic enterococcus faecium attenuated atherosclerosis by improving SCFAs associated with gut microbiota in ApoE-/- mice. Bioengineering. 2024;11:1033.
126. Yang H, Yang Y, Cui G, et al. Dietary methionine restriction ameliorates atherosclerosis by remodeling the gut microbiota in apolipoprotein E-knockout mice. Food Funct. 2025;16:4904-22.
127. Luo Z, Yang L, Zhu T, et al. Aucubin ameliorates atherosclerosis by modulating tryptophan metabolism and inhibiting endothelial-mesenchymal transitions via gut microbiota regulation. Phytomedicine. 2024;135:156122.
128. Nageswaran V, Carreras A, Reinshagen L, et al. Gut microbial metabolite imidazole propionate impairs endothelial cell function and promotes the development of atherosclerosis. Arterioscler Thromb Vasc Biol. 2025;45:823-39.
129. Paone S, Baxter AA, Hulett MD, Poon IKH. Endothelial cell apoptosis and the role of endothelial cell-derived extracellular vesicles in the progression of atherosclerosis. Cell Mol Life Sci. 2019;76:1093-106.
130. Lin W, Huang F, Yuan Y, et al. Endothelial exosomes work as a functional mediator to activate macrophages. Front Immunol. 2023;14:1169471.
131. He S, Wu C, Xiao J, Li D, Sun Z, Li M. Endothelial extracellular vesicles modulate the macrophage phenotype: Potential implications in atherosclerosis. Scand J Immunol. 2018;87:e12648.
132. Chang YJ, Li YS, Wu CC, et al. Extracellular MicroRNA-92a mediates endothelial cell-macrophage communication. Arterioscler Thromb Vasc Biol. 2019;39:2492-504.
133. Bennett MR, Sinha S, Owens GK. Vascular smooth muscle cells in atherosclerosis. Circ Res. 2016;118:692-702.
134. Wang H, Xie Y, Salvador AM, et al. Exosomes: multifaceted messengers in atherosclerosis. Curr Atheroscler Rep. 2020;22:57.
135. Zhang Z, Yi D, Zhou J, et al. Exosomal LINC01005 derived from oxidized low-density lipoprotein-treated endothelial cells regulates vascular smooth muscle cell phenotypic switch. Biofactors. 2020;46:743-53.
136. Dabravolski SA, Sukhorukov VN, Glanz VY, Pleshko EM, Orekhov NA, Sobenin IA. Exosomes in atherosclerosis: role in the pathogenesis and targets for therapy. Curr Med Chem. 2025;32:3106-21.
137. Xu L, Geng T, Zang G, et al. Exosome derived from CD137-modified endothelial cells regulates the Th17 responses in atherosclerosis. J Cell Mol Med. 2020;24:4659-67.
138. Gao H, Wang X, Lin C, et al. Exosomal MALAT1 derived from ox-LDL-treated endothelial cells induce neutrophil extracellular traps to aggravate atherosclerosis. Biol Chem. 2020;401:367-76.
139. Chen L, Hu L, Li Q, Ma J, Li H. Exosome-encapsulated miR-505 from ox-LDL-treated vascular endothelial cells aggravates atherosclerosis by inducing NET formation. Acta Biochim Biophys Sin. 2019;51:1233-41.
140. Liu P, Wang S, Wang G, et al. Macrophage-derived exosomal miR-4532 promotes endothelial cells injury by targeting SP1 and NF-κB P65 signalling activation. J Cell Mol Med. 2022;26:5165-80.
141. Tang N, Sun B, Gupta A, Rempel H, Pulliam L. Monocyte exosomes induce adhesion molecules and cytokines via activation of NF-κB in endothelial cells. FASEB J. 2016;30:3097-106.
142. Chen S, Zhou H, Zhang B, Hu Q. Exosomal miR-512-3p derived from mesenchymal stem cells inhibits oxidized low-density lipoprotein-induced vascular endothelial cells dysfunction via regulating Keap1. J Biochem Mol Toxicol. 2021;35:1-11.
143. Xing X, Li Z, Yang X, et al. Adipose-derived mesenchymal stem cells-derived exosome-mediated microRNA-342-5p protects endothelial cells against atherosclerosis. Aging. 2020;12:3880-98.
144. Li J, Tan M, Xiang Q, Zhou Z, Yan H. Thrombin-activated platelet-derived exosomes regulate endothelial cell expression of ICAM-1 via microRNA-223 during the thrombosis-inflammation response. Thromb Res. 2017;154:96-105.
145. Segal SS, Bagher P. Regulation of myoendothelial junction formation: bridging the gap. Circ Res. 2010;106:1014-6.
146. Shu X, Ruddiman CA, Keller TCS 4th, et al. Heterocellular contact can dictate arterial function. Circ Res. 2019;124:1473-81.
147. Lemmey HA, Garland CJ, Dora KA. Intrinsic regulation of microvascular tone by myoendothelial feedback circuits. Curr Top Membr. 2020;85:327-55.
148. Straub AC, Zeigler AC, Isakson BE. The myoendothelial junction: connections that deliver the message. Physiology. 2014;29:242-9.
149. Ledoux J, Taylor MS, Bonev AD, et al. Functional architecture of inositol 1,4,5-trisphosphate signaling in restricted spaces of myoendothelial projections. Proc Natl Acad Sci U S A. 2008;105:9627-32.
150. Han Y, Huang K, Yao Q, Jiang Z. Mechanobiology in vascular remodeling. Nat Sci Rev. 2018;5:933-46.
151. Lateef OM, Foote CA, Ramirez-perez FI, et al. Vascular smooth muscle cell mechanical stretch modulates tissue transglutaminase activity and cytoskeletal dynamics. Physiology. 2024;39:735.
152. Zaric B, Obradovic M, Trpkovic A, Banach M, Mikhailidis DP, Isenovic ER. Endothelial dysfunction in dyslipidaemia: molecular mechanisms and clinical implications. Curr Med Chem. 2020;27:1021-40.
153. Gao D, Hua R, Jiesisibieke D, et al. C-reactive protein and coronary atheroma regression following statin therapy: a meta-regression of randomized controlled trials. Front Cardiovasc Med. 2022;9:989527.
154. Egede R, Jensen LO, Hansen HS, et al. Effect of intensive lipid-lowering treatment compared to moderate lipid-lowering treatment with rosuvastatin on endothelial function in high risk patients. Int J Cardiol. 2012;158:376-9.
155. Takayama T, Hiro T, Yoda S, et al. Effect of aggressive lipid-lowering treatment with rosuvastatin on vascular endoTHelium function: evaluation of vascular endothelium function (EARTH study). Heart Vessels. 2018;33:590-4.
156. Kater AL, Batista MC, Ferreira SR. Improved endothelial function with simvastatin but unchanged insulin sensitivity with simvastatin or ezetimibe. Metabolism. 2010;59:921-6.
157. Ziogos E, Chelko SP, Harb T, et al. Platelet activation and endothelial dysfunction biomarkers in acute coronary syndrome: the impact of PCSK9 inhibition. Eur Heart J Cardiovasc Pharmacother. 2023;9:636-46.
158. Liu S, Wu J, Stolarz A, et al. PCSK9 attenuates efferocytosis in endothelial cells and promotes vascular aging. Theranostics. 2023;13:2914-29.
159. Taddei S, Virdis A, Ghiadoni L, Sudano I, Salvetti A. Effects of antihypertensive drugs on endothelial dysfunction: clinical implications. Drugs. 2002;62:265-84.
160. Miyamoto M, Kotani K, Ishibashi S, Taniguchi N. The effect of antihypertensive drugs on endothelial function as assessed by flow-mediated vasodilation in hypertensive patients. Int J Vasc Med. 2012;2012:453264.
161. Flammer AJ, Gössl M, Li J, et al. Renin inhibition with aliskiren lowers circulating endothelial progenitor cells in patients with early atherosclerosis. J Hypertens. 2013;31:632-5.
162. Virdis A, Ghiadoni L, Taddei S. Effects of antihypertensive treatment on endothelial function. Curr Hypertens Rep. 2011;13:276-81.
163. Wang Y, Yao M, Wang J, et al. Effects of antidiabetic drugs on endothelial function in patients with type 2 diabetes mellitus: a bayesian network meta-analysis. Front Endocrinol. 2022;13:818537.
164. Maruhashi T, Higashi Y. Pathophysiological association between diabetes mellitus and endothelial dysfunction. Antioxidants. 2021;10:1306.
165. Dandona P, Aljada A. Endothelial dysfunction in patients with type 2 diabetes and the effects of thiazolidinedione antidiabetic agents. J Diabetes Complications. 2004;18:91-102.
166. Pereira CA, Carneiro FS, Matsumoto T, Tostes RC. Bonus effects of antidiabetic drugs: possible beneficial effects on endothelial dysfunction, vascular inflammation and atherosclerosis. Basic Clin Pharmacol Toxicol. 2018;123:523-38.
167. Huang AL, Vita JA. Effects of systemic inflammation on endothelium-dependent vasodilation. Trends Cardiovasc Med. 2006;16:15-20.
168. Shao Y, Cheng Z, Li X, Chernaya V, Wang H, Yang XF. Immunosuppressive/anti-inflammatory cytokines directly and indirectly inhibit endothelial dysfunction-a novel mechanism for maintaining vascular function. J Hematol Oncol. 2014;7:80.
169. Vera M, Torramade-Moix S, Martin-Rodriguez S, et al. Antioxidant and anti-inflammatory strategies based on the potentiation of glutathione peroxidase activity prevent endothelial dysfunction in chronic kidney disease. Cell Physiol Biochem. 2018;51:1287-300.
170. Penna C, Pagliaro P. Endothelial dysfunction: redox imbalance, NLRP3 inflammasome, and inflammatory responses in cardiovascular diseases. Antioxidants. 2025;14:256.
171. van der Heijden T, Kritikou E, Venema W, et al. NLRP3 Inflammasome inhibition by MCC950 reduces atherosclerotic lesion development in apolipoprotein E-deficient mice-brief report. Arterioscler Thromb Vasc Biol. 2017;37:1457-61.
172. Kajikawa M, Higashi Y, Tomiyama H, et al. Effect of short-term colchicine treatment on endothelial function in patients with coronary artery disease. Int J Cardiol. 2019;281:35-9.
173. Tanase DM, Valasciuc E, Gosav EM, et al. Portrayal of NLRP3 inflammasome in atherosclerosis: current knowledge and therapeutic targets. Int J Mol Sci. 2023;24:8162.
174. Mankan AK, Dau T, Jenne D, Hornung V. The NLRP3/ASC/Caspase-1 axis regulates IL-1β processing in neutrophils. Eur J Immunol. 2012;42:710-5.
175. Paget C, Doz-Deblauwe E, Winter N, Briard B. Specific NLRP3 inflammasome assembling and regulation in neutrophils: relevance in inflammatory and infectious diseases. Cells. 2022;11:1188.
176. Fountoulakis PN, Theofilis P, Vlachakis PK, et al. Gut microbiota in heart failure-the role of inflammation. Biomedicines. 2025;13:911.
177. Dufour D, Khalil A, Nuyens V, et al. Native and myeloperoxidase-oxidized low-density lipoproteins act in synergy to induce release of resolvin-D1 from endothelial cells. Atherosclerosis. 2018;272:108-17.
178. Pichavaram P, Mani AM, Singh NK, Rao GN. Cholesterol crystals promote endothelial cell and monocyte interactions via H2O2-mediated PP2A inhibition, NFκB activation and ICAM1 and VCAM1 expression. Redox Biol. 2019;24:101180.
179. Varadharaj S, Kelly OJ, Khayat RN, Kumar PS, Ahmed N, Zweier JL. Role of dietary antioxidants in the preservation of vascular function and the modulation of health and disease. Front Cardiovasc Med. 2017;4:64.
180. Mortensen A, Lykkesfeldt J. Does vitamin C enhance nitric oxide bioavailability in a tetrahydrobiopterin-dependent manner? Nitric Oxide. 2014;36:51-7.
181. Paulo M, Costa DEFR, Bonaventura D, Lunardi CN, Bendhack LM. Nitric oxide donors as potential drugs for the treatment of vascular diseases due to endothelium dysfunction. Curr Pharm Des. 2020;26:3748-59.
182. Parsamanesh N, Asghari A, Sardari S, et al. Resveratrol and endothelial function: a literature review. Pharmacol Res. 2021;170:105725.
183. Ulker S, McKeown PP, Bayraktutan U. Vitamins reverse endothelial dysfunction through regulation of eNOS and NAD(P)H oxidase activities. Hypertension. 2003;41:534-9.
184. Xue HM, Yu CM, Underwood MJ, Huang JH, Yang Q. AVE3085 protects coronary endothelium from the impairment of asymmetric dimethylarginine by activation and recoupling of eNOS. Cardiovasc Drugs Ther. 2012;26:383-92.
185. Yang Q, Xue HM, Underwood MJ, Yu CM. Mechanistic studies of AVE3085 against homocysteine in endothelial protection. Cardiovasc Drugs Ther. 2013;27:511-20.
186. Yang Q, Xue HM, Wong WT, et al. AVE3085, an enhancer of endothelial nitric oxide synthase, restores endothelial function and reduces blood pressure in spontaneously hypertensive rats. Br J Pharmacol. 2011;163:1078-85.
187. Li X, Zou J, Lin A, et al. Oxidative stress, endothelial dysfunction, and N-acetylcysteine in type 2 diabetes mellitus. Antioxid Redox Signal. 2024;40:968-89.
188. Vera R, Sánchez M, Galisteo M, et al. Chronic administration of genistein improves endothelial dysfunction in spontaneously hypertensive rats: involvement of eNOS, caveolin and calmodulin expression and NADPH oxidase activity. Clin Sci. 2007;112:183-91.
189. Pottecher J, Cheisson G, Huet O, et al. β2-adrenergic agonist protects human endothelial cells from hypoxia/reoxygenation injury in vitro. Crit Care Med. 2006;34:165-72.
190. Barbato E, Herman A, Benit E, et al. Long-term effect of molsidomine, a direct nitric oxide donor, as an add-on treatment, on endothelial dysfunction in patients with stable angina pectoris undergoing percutaneous coronary intervention: results of the MEDCOR trial. Atherosclerosis. 2015;240:351-4.
191. Vera OD, Wulff H, Braun AP. Endothelial KCa channels: novel targets to reduce atherosclerosis-driven vascular dysfunction. Front Pharmacol. 2023;14:1151244.
192. Gao F, Chen J, Zhu H. A potential strategy for treating atherosclerosis: improving endothelial function via AMP-activated protein kinase. Sci China Life Sci. 2018;61:1024-9.
193. Tong X, Dang X, Liu D, et al. Exosome-derived circ_0001785 delays atherogenesis through the ceRNA network mechanism of miR-513a-5p/TGFBR3. J Nanobiotechnology. 2023;21:362.
194. Huang HC, Wang TY, Rousseau J, et al. Biomimetic nanodrug targets inflammation and suppresses YAP/TAZ to ameliorate atherosclerosis. Biomaterials. 2024;306:122505.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0










Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].