


Advancing atherosclerosis research: mouse models as tools for translational applications
 0
0 Abstract
Atherosclerosis research has been significantly advanced by mouse models, particularly genetically engineered strains such as apolipoprotein E-deficient mice and low-density lipoprotein receptor-deficient mice (Ldlr-/-). These mouse models replicate the hyperlipidemia-driven plaque pathogenesis, providing critical insights into lipid metabolism, inflammation, and therapeutic responses. Classic models play an important role in validating the effects of lipid-lowering therapies such as statins and proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors. There are also some limitations, including species-specific lipoprotein profiles and incomplete replication of advanced human plaque complexity. Novel models have emerged to address these gaps, incorporating features such as hemodynamic stress, humanized lipid metabolism, and inducible gene regulation to complement the inadequacies of classic models, thereby better simulating the multifactorial complexity of atherosclerosis. For example, adeno-associated virus serotype 8 carrying the Pcsk9[D377Y] mutant gene and Ldlr-antisense oligonucleotide mice integrate hemodynamic stress, humanized lipid metabolism, and multifactorial comorbidities. However, there are still some shortcomings, including metabolic disparities, inadequate modeling of plaque rupture/thrombosis, and oversimplification of systemic disease interactions. Future directions prioritize next-generation models featuring humanized lipoprotein profiles, dynamic gene regulation, and combined with metabolic syndrome features. These approaches can be synergized with advanced phenotyping tools—including single-cell omics and intravital imaging—alongside artificial intelligence-driven multi-omics integration. By bridging translational gaps between murine pathophysiology and human disease complexity, these mouse models promise to accelerate the development of atherosclerosis therapies.
Keywords
INTRODUCTION
Atherosclerosis, a chronic inflammatory disease of the arterial wall, remains a leading cause of global morbidity and mortality through its serious complications—myocardial infarction, stroke, and peripheral artery disease[1-3]. It is characterized by lipid-rich plaque formation, endothelial dysfunction, and immune-mediated vascular remodeling[4]. This complex pathology results from intricate interactions among genetic predisposition, metabolic disturbances, inflammation, and environmental triggers[5,6]. For decades, research has elucidated key mechanisms of atherosclerosis, spanning from oxidized low-density lipoprotein (LDL)-driven foam cell formation to cytokine-mediated plaque instability[7,8]. Translating preclinical discoveries into effective clinical therapies remains a significant challenge, despite this progress. This translational gap highlights the critical need for experimental models capable of mimicking human disease dynamics.
Mouse models have served as indispensable tools in atherosclerosis research since the landmark development of apoprotein E (ApoE)-deficient and Ldl receptor knockout strains in the 1990s[9,10]. These models bridge the gaps between in vitro studies and human trials by replicating key features of hyperlipidemia-driven atherogenesis[11,12]. Furthermore, translational relevance has been strengthened by subsequent refinement. This includes models incorporating human transgenes and multifactorial disease drivers such as endothelial dysfunction and metabolic syndrome[13]. However, there are still some limitations in mouse models, including species-specific lipoprotein metabolism and incomplete replication of advanced plaque complications[14].
This review provides a comprehensive analysis of the evolving landscape of mouse atherosclerosis models. We first describe the foundational role of classic models—specifically apolipoprotein E-deficient mice (ApoE-/-) and LDL receptor-deficient mice (Ldlr-/-)—in establishing causal relationships between dyslipidemia and plaque biology. Concurrently, we evaluate their strengths and inherent biological limitations. Subsequent sections elaborate on innovative mouse models addressing these limitations, such as apolipoprotein E3 Leiden cholesteryl ester transfer protein (ApoE3-Leiden.CETP) mice, Ldlr-antisense oligonucleotide (ASO) models, and Scavenger receptor class B type I C-terminal truncation mutant (SR-BIΔCT/ΔCT)/Ldlr-/- mice. A dedicated discussion of metabolic disparities and pathological simplification in current models highlights the urgent need for next-generation models. This review aims to inform model selection strategies and inspire novel mouse models to bridge the persistent divide between laboratory discoveries and clinical applications.
ATHEROSCLEROSIS AND THE ROLE OF MOUSE MODELS
Atherosclerosis is a chronic inflammatory disease of the arterial wall. It is initiated by endothelial injury due to hemodynamic stress, hyperlipidemia, or oxidative stress, leading to the recruitment of monocytes and their differentiation into macrophages[15,16]. A pivotal early event in this process is the retention of LDL within the subendothelial intima[17,18], which represents a key mechanism in atherosclerosis development. Following endothelial dysfunction, circulating LDL particles infiltrate the arterial wall and, once within the intima, bind via positively charged residues of apolipoprotein B-100 to negatively charged proteoglycans in the extracellular matrix[19]. This electrostatic interaction traps LDL, prolonging its residence time and increasing its concentration in the vascular wall[20]. The retained LDL then becomes susceptible to a range of modifications, including oxidation, aggregation, and enzymatic degradation. These modifications, particularly oxidation, generate pro-inflammatory lipids and neo-epitopes that are recognized by scavenger receptors on macrophages[21]. This markedly enhances the uptake of modified LDL and drives the transformation of macrophages into lipid-laden foam cells. Thus, LDL retention acts as a critical trigger that converts transient endothelial permeability into a sustained pathological accumulation of atherogenic lipoproteins. These macrophages engulf oxidized LDL, forming foam cells that drive plaque formation[22-24]. Proinflammatory cytokines and chemokines further amplify leukocyte recruitment and smooth muscle cell proliferation, resulting in fibrous cap development and plaque instability[25]. If left uncontrolled, advanced plaques may rupture, causing thrombosis, myocardial infarction, or stroke[26,27]. Advances in understanding disease mechanisms have been significant. Nevertheless, translating preclinical findings into clinical therapies remains challenging, necessitating robust experimental models to dissect disease and test interventions [Figure 1].
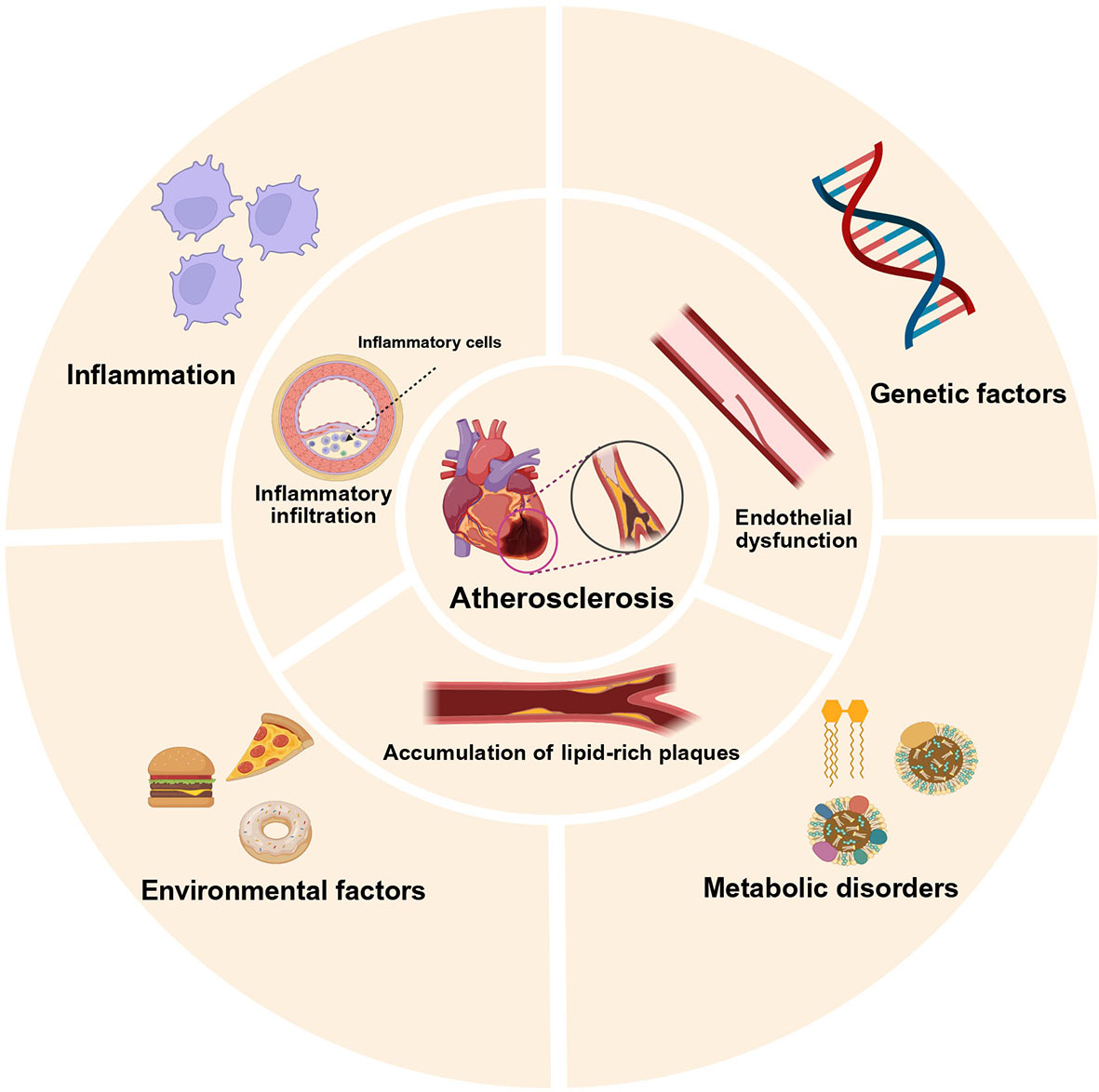
Figure 1. Factors and characteristics of atherosclerosis. The inner ring depicts pathological features of atherosclerosis: lipid-rich plaques, endothelial dysfunction, and immune cell infiltration; the outer ring represents contributing risk factors: genetic, metabolic, inflammation and environmental factors. Created in BioRender. kaisi, Y. (2025) https://BioRender.com/mc3lp37.
Mouse models have become indispensable tools in atherosclerosis research, bridging the gap between cellular studies and human trials. While in vitro systems provide mechanistic insights, they lack the complexity of systemic interactions in vivo[28]. Human studies are constrained by ethical limitations and genetic heterogeneity, although they remain indispensable for research. In this context, genetically engineered mouse models have played a crucial role in atherosclerosis research. They replicate key features of human atherosclerosis, including hyperlipidemia-driven plaque development and inflammation[29,30]. Furthermore, these models enable precise manipulation of genes, diets, and therapeutic agents, facilitating the exploration of causal relationships and therapeutic targets[31]. They mimic atherosclerosis progression and regression, serving as translational tools to evaluate novel drugs and gene therapies. This capability underscores their irreplaceable role in advancing atherosclerosis research from laboratory discovery to clinical application.
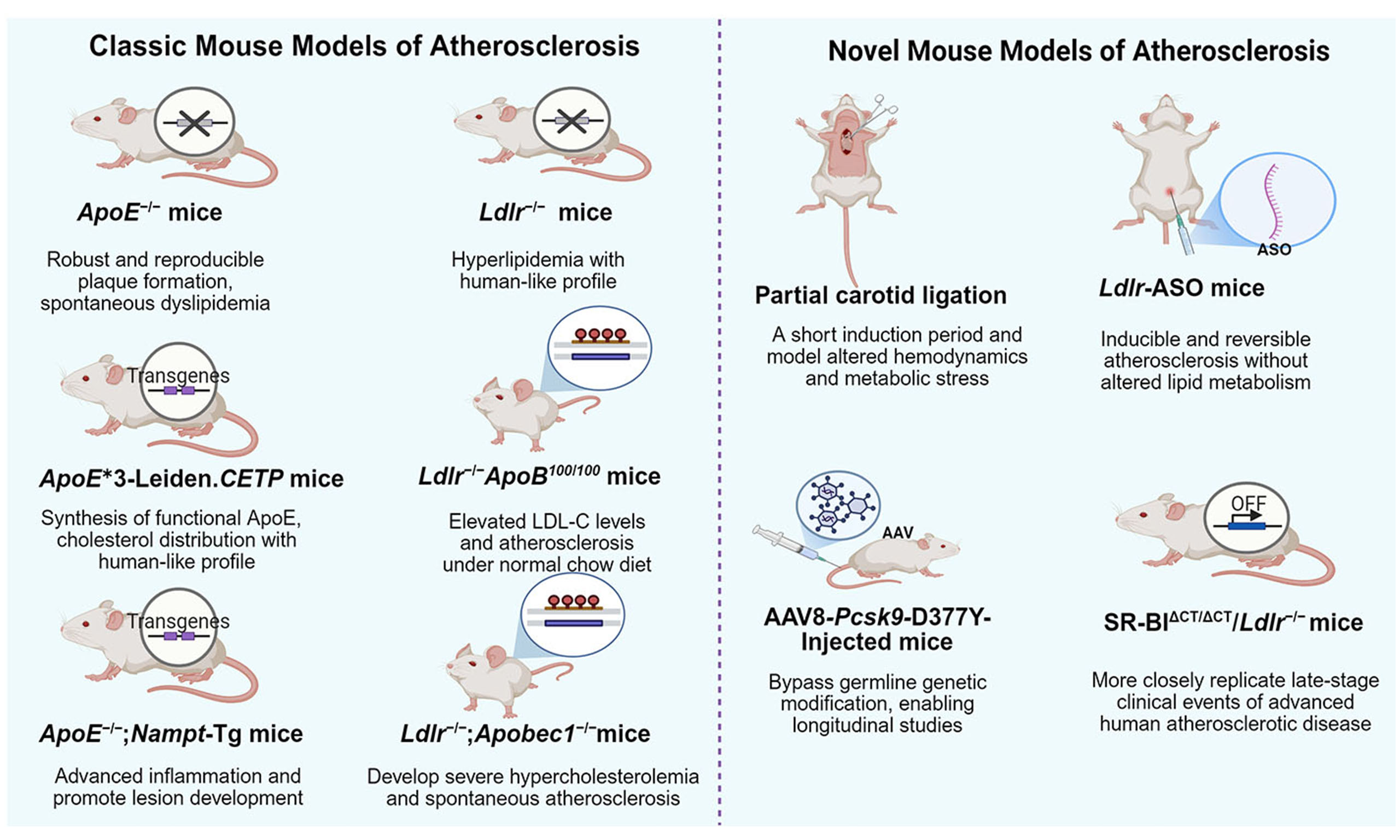
CLASSIC MOUSE MODELS OF ATHEROSCLEROSIS: CONSTRUCTION STRATEGIES AND TRANSLATIONAL APPLICATIONS
Classic mouse models, notably ApoE-/- and Ldlr-/- strains, have been pivotal in atherosclerosis research[32,33]. They enable foundational insights into lipid metabolism, inflammation, and therapeutic responses to agents such as statins and proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors[34]. These models also have some limitations, such as species-specific lipoprotein profiles dominated by very low-density lipoprotein (VLDL) and chylomicrons in mice and compensatory metabolic adaptation[35,36]. These constraints have driven the development of advanced strains. Enhanced models address these gaps through incorporating human transgenes and multi-gene perturbations, such as ApoE3-Leiden.CETP and Ldlr-/-ApoB100/100 strains. These strategic refinements improve translational relevance for studying dyslipidemia, endothelial dysfunction, and patient-specific mutations[37,38]. Collectively, both classic and refined models are indispensable for bridging preclinical discoveries to clinical applications [Table 1].
Classic and novel mouse models of atherosclerosis
| Mouse models of atherosclerosis | The strategies of modeling | Advantages | Limitations | Translational applications | References |
| Classic mouse models | |||||
| ApoE-/- mice | Normal chow/HFD | Robust and reproducible plaque formation; spontaneous dyslipidemia | Fail to accurately replicate human lipid profiles; rare spontaneous plaque rupture | Test therapies targeting lipid metabolism, inflammation, and oxidative stress | [32,39] |
| ApoE ∗3-Leiden.CETP mice | HFD | Synthesis of functional ApoE; no effect on inflammation; human-like cholesterol distribution | Significant inter-individual variations | Evaluate lipid-modifying pharmacotherapies | [49,50] |
| ApoE-/-; Nampt-Tg mice | HFD | Significantly increased lesion area, thickness, and inflammation | Rare occurrence of plaque rupture and thrombosis | Study of inflammation in atherosclerosis | [53] |
| Ldlr-/- mice | Normal chow/HFD | Hyperlipidemia with human-like profiles | Requires prolonged HFD feeding; no coronary lesion formation | Targeting familial hypercholesterolemia-like LDLR deficiency | [54,56] |
| Ldlr-/-ApoB100/100 mice | Normal chow | Elevated LDL-C and atherosclerosis on normal chow; human-like lipid-rich plaques | Absence of spontaneous plaque rupture | Investigate LDL-C-driven inflammation and evaluating anti-inflammatory therapies | [62,63] |
| Ldlr-/-;Apobec1-/- mice | Normal chow | Develops severe hypercholesterolemia and spontaneous atherosclerosis on normal chow | Prolonged time course for lesion development | Suitable for testing novel anti-atherosclerotic drugs | [65,67] |
| Novel mouse models | |||||
| Partial artery ligation combined with HFD | Surgery + HFD | Short induction period; integrate hemodynamic stress and metabolic dysregulation | Lack plaque rupture, diverging from advanced human atherosclerosis | Identify potential therapeutic interventions for disturbed blood flow-induced endothelial dysfunction | [77,78] |
| Ldlr-ASO mice | ASO injection + HFD | Inducible and reversible atherosclerosis without altered lipid metabolism | Depend on repeated ASO dosing | Provide a valuable platform for evaluating anti-atherosclerotic drugs | [86,87] |
| AAV8-Pcsk9-D377Y-Injected mice | AAV8-Pcsk9 injection + HFD | Bypass germline genetic modification, enabling longitudinal studies | Fail to replicate spontaneous plaque rupture; exhibit inter-individual variability | Rapidly inducing atherosclerosis and identifying regulators of atherogenesis | [94,96] |
| WD-Fed SR-BI∆CT/∆CT/Ldlr-/- mice | Western diet | Closely replicate late-stage clinical events of advanced human atherosclerotic disease | Require prolonged disease induction | Test potential new therapeutic agents to prevent major adverse cardiac events | [73] |
ApoE-/- mice
ApoE-/- mouse model, first described in the early 1990s, is one of the most widely used models in atherosclerosis research[39]. ApoE is a key regulator of lipid metabolism, mediating the clearance of VLDL and chylomicrons via binding to LDL receptors in mice[40]. In ApoE-/- mice, the ApoE gene is permanently inactivated through homologous recombination. This genetic modification results in impaired lipoprotein clearance, spontaneous hypercholesterolemia, and accelerated atherosclerosis[13]. These mice develop lipid-laden atherosclerotic plaques even on a standard chow diet, though plaque progression is markedly accelerated by a high-fat diet (HFD)[32]. Histologically, lesions in ApoE-/- mice reproduce human plaque features, including foam cell accumulation, necrotic cores, and fibrous caps[41].
Thestrengths of ApoE-/- mice lie in their robust and reproducible plaque formation, and utility for studying both early and advanced atherosclerosis[42]. Their spontaneous dyslipidemia eliminates the need for chemical or surgical interventions to induce disease. Additionally, their compatibility with bone marrow transplantation enables studies on hematopoietic cell-specific contributions to atherosclerosis[41].
However, ApoE-/- mice have important limitations. They transport the majority of plasma cholesterol via VLDL and chylomicron remnants, whereas LDL is the primary cholesterol carrier in humans[43]. Furthermore, lesions in ApoE-/- mice rarely rupture and do not lead to thrombosis, whereas vascular occlusion is common in humans[44]. Despite these disadvantages, their translational relevance is supported by their responsiveness to lipid-lowering therapies, such as statins and PCSK9 inhibitors.
ApoE-/- mice have been pivotal in preclinical drug discovery. For example, they play an important role in validating the efficacy of statins, which reduce plaque burden by lowering LDL cholesterol and suppressing vascular inflammation[45]. Studies using these models also reveal the anti-atherogenic effects of PCSK9 inhibitors, which enhance low-density lipoprotein receptor (LDLR) recycling and reduce circulating LDL levels[46]. Beyond lipid-modulating therapies, ApoE-/- mice have facilitated testing of novel agents targeting inflammatory pathways such as interleukin (IL)-1β inhibition and oxidative stress mediators, including nicotinamide adenine dinucleotide phosphate oxidase inhibitors[47,48]. ApoE-/- mice effectively mirror therapeutic responses observed in human clinical trials. These abilities establish their essential value in bridging preclinical and clinical research.
Other genetically modified ApoE-/- mouse strains
ApoE3-Leiden.CETP mice
The ApoE3-Leiden.CETP mouse model is engineered by hybridizing ApoE3-Leiden mice with transgenic mice expressing human CETP[49]. Unlike ApoE-/- models, this strain retains native ApoE synthesis. This retention avoids artificial disruption of inflammatory pathways in atherosclerotic lesions while enabling precise modulation of plasma lipid profiles. The ApoE3-Leiden transgene impairs clearance of triglyceride-rich lipoproteins such as VLDL remnants and chylomicrons, whereas CETP overexpression shifts cholesterol distribution toward a human-like pattern, particularly under HFD conditions[50]. Consequently, these mice develop mixed hyperlipidemia (elevated VLDL/LDL) and progressive atherosclerosis. However, upon HFD feeding, this model develops metabolic syndrome and exhibits significant inter-individual variations despite the absence of genetic or environmental diversity[51].
This strain is extensively employed to evaluate lipid-modifying pharmacotherapies such as statins, fibrates, and CETP inhibitors[52]. Its humanized lipoprotein metabolism significantly enhances the translational relevance for studying CETP-targeted therapeutic strategies.
ApoE-/-;Nampt-Tg mice
The ApoE-/-;nicotinamide phosphoribosyltransferase (Nampt-Tg) strain combines ApoE deficiency with transgenic overexpression of Nampt, a rate-limiting enzyme in nicotinamide adenine dinucleotide biosynthesis[53]. Lesion area, thickness, and inflammatory markers of atherosclerosis are significantly increased in ApoE-/-;Nampt-Tg mice compared with ApoE-/- mice after 16 weeks of HFD feeding[53]. However, a critical limitation is the model's inability to mimic acute cardiovascular events, due to rare occurrences of plaque rupture and subsequent thrombosis.
Despite limited current utilization in atherosclerosis studies, this model offers a unique way to dissect the crosstalk of metabolic reprogramming and inflammatory signaling cascades underlying the pathology.
Ldlr-/- mice
The Ldlr-/- mouse model is also a cornerstone of atherosclerosis research. This model is generated through homologous recombination to disrupt the Ldlr gene[54]. LDLR plays a critical role in hepatic clearance of circulating LDL by mediating endocytosis of LDL particles[55]. In Ldlr-/- mice, the absence of functional LDLR leads to impaired LDL catabolism, resulting in elevated plasma LDL-cholesterol (LDL -C) levels. Ldlr-/- mice display low-grade atherosclerotic phenotypes when fed a normal chow, but switching to an HFD robustly elevates circulating lipids and intensifies atherosclerosis severity[56]. This diet-induced phenotype allows researchers to modulate disease progression temporally, making the model ideal for studying interventions targeting lipid metabolism. Atherosclerotic lesions in Ldlr-/- mice predominantly localize to the aortic root and arch. These lesions demonstrate histopathological features mirroring human plaques, with lipid-rich cores, macrophage infiltration, and fibrous cap formation[13].
The Ldlr-/- model offers precise control over hyperlipidemia and plaque development through dietary manipulation. Its genetic tractability facilitates the generation of compound mutants (e.g., Ldlr-/-; apolipoprotein B100 (ApoB100) transgenic mice) to mimic human-like lipoprotein profiles[57]. Additionally, Ldlr-/- mice exhibit robust responsiveness to lipid-lowering therapies, such as statins and ezetimibe, enhancing their utility in pharmacodynamic studies[58].
A key limitation of Ldlr-/- mice is the requirement for prolonged HFD feeding (12-24 weeks) to induce advanced plaques[59]. This requirement significantly extends experimental timelines. Furthermore, Ldlr-/- mice predominantly develop atherosclerosis in the aortic root rather than the coronary arteries, diverging from human disease patterns[13]. The model remains highly relevant for studying LDL-C-driven atherosclerosis, despite these constraints.
The Ldlr-/- mouse model serves as an indispensable translational bridge in cardiovascular research. It enables mechanistic dissection of LDL-C-driven atherosclerosis and therapeutic validation for familial hypercholesterolemia. Its precision in mimicking impaired LDL clearance continues to accelerate the development of targeted agents—from PCSK9 inhibitors to next-generation therapies[60,61]. These capacities solidify the model's translational relevance in cardiometabolic drug discovery.
Other genetically modified Ldlr-/- mouse strains
Ldlr-/- ApoB100/100 mice
The Ldlr-/-ApoB100/100 model is generated by introducing a human apolipoprotein B (ApoB) transgene into Ldlr-/- mice, replacing murine ApoB48 with human ApoB100[62]. This modification mimics human-like LDL metabolism, as ApoB100 serves as the sole structural apolipoprotein on LDL particles in humans. These mice exhibit elevated LDL-C levels and develop lipid-rich atherosclerotic plaques under a normal chow diet[62]. These plaques have histopathological features resembling human lesions. However, a critical limitation is the absence of spontaneous plaque rupture, restricting this model’s utility for studying advanced thrombotic complications[63].
Despite this constraint, the model remains pivotal for investigating LDL-C-driven inflammation and evaluating anti-inflammatory therapies. This model is used to study the various effects of statins, such as suppressing pro-inflammatory cytokine release and attenuating macrophage infiltration in plaques[64]. These findings directly inform clinical strategies for stabilizing vulnerable plaques via statin-mediated inflammation modulation.
Ldlr-/-;Apobec1-/- mice
APOB messenger RNA (mRNA)-editing enzyme catalytic subunit 1 (Apobec1) is responsible for post-transcriptional editing of APOB mRNA to generate ApoB48 in rodents[65]. In Ldlr-/-;Apobec1-/- mice, the absence of Apobec1 prevents ApoB48 production, forcing exclusive expression of ApoB100. This model develops severe hypercholesterolemia and spontaneous atherosclerosis even on a chow diet[66]. This eliminates the requirement for dietary manipulation. However, the pathological progression in this model involves a prolonged and complex timeline, which extends the experimental period[67]. This strain is suitable for testing the effects of new potential anti-atherosclerotic drugs.
Mechanistic basis for selecting ApoE-/- or Ldlr-/- mouse models in atherosclerosis research
The distinct molecular etiologies of hypercholesterolemia in ApoE-/- and Ldlr-/- mice are the basis of their respective applications in atherosclerosis research. This divergence in mechanism determines the specific atherogenic lipoprotein profile, thus providing appropriate model selection for different research questions. ApoE-/- mice develop severe hypercholesterolemia primarily due to the loss of ApoE-mediated clearance of triglyceride-rich lipoprotein remnants, such as chylomicron remnants and VLDL remnants[68,69]. Consequently, their plasma is dominated by cholesterol-enriched remnant lipoproteins, modeling disorders of remnant metabolism. In contrast, hypercholesterolemia in Ldlr-/- mice results from the direct ablation of the hepatic LDLR pathway, which is responsible for the endocytic clearance of circulating LDL particles[69,70]. This leads to a pronounced accumulation of ApoB100-containing LDL, making it a more direct model for LDL-C-driven pathogenesis, such as familial hypercholesterolemia. Therefore, although both models produce elevated plasma cholesterol and atherosclerotic plaque, their underlying pathophysiology is fundamentally different. Specifically, ApoE-/- mice are particularly suited for studying remnant lipoprotein biology and associated inflammation[71]. Ldlr-/- mice are the model of choice for investigating LDL-driven pathways and therapies targeting the LDLR axis, such as PCSK9 inhibitors[71,72]. Understanding this core molecular distinction is crucial for designing experiments that accurately mimic specific facets of human dyslipidemia.
NOVEL MOUSE MODELS OF ATHEROSCLEROSIS: CONSTRUCTION STRATEGIES AND TRANSLATIONAL APPLICATIONS
Emerging mouse models of atherosclerosis address limitations of classic genetic models by integrating hemodynamic, pharmacological, and multifactorial dyslipidemia approaches[9,73]. The partial carotid ligation (PCL) + HFD model combines mechanical endothelial injury with metabolic stress[74]. This combination enables rapid plaque induction and studies on hemodynamic-inflammatory crosstalk. Ldlr-ASO mice utilize reversible, hepatocyte-specific Ldlr suppression to model acquired hypercholesterolemia[75]. adeno-associated virus serotype 8 carrying the Pcsk9[D377Y] mutant gene (AAV8-Pcsk9-D377Y) mice achieve stable hepatic Pcsk9 overexpression, mirroring familial hypercholesterolemia and accelerating PCSK9-targeted drug discovery[76]. The SR-BI∆CT/∆CT/Ldlr-/- model uniquely mimics major adverse cardiovascular events, including spontaneous plaque rupture with myocardial infarction and stroke[73]. The SR-BI∆CT/∆CT/Ldlr-/- model can be used to identify and test potential new therapeutic agents to prevent major adverse cardiac events. Collectively, these innovative models enhance translational relevance by incorporating human-like pathophysiology, such as coronary lesions and plaque regression.
Partial carotid ligation (PCL) combined with HFD
The PCL combined with HFD is a surgically induced atherosclerosis model designed to study plaque formation under conditions of altered hemodynamics and metabolic stress[77,78]. In this model, unilateral or bilateral ligation of the common carotid artery is performed surgically, leading to reduced blood flow and low shear stress in the ligated segment. This hemodynamic perturbation triggers endothelial dysfunction, monocyte recruitment, and accelerated plaque development[79]. When combined with HFD feeding, systemic hyperlipidemia exacerbates vascular inflammation and lipid deposition at the ligation site. Atherosclerotic lesions in this model develop within 2-4 weeks, characterized by intimal hyperplasia, macrophage infiltration, and neointima formation[80].
This model enables rapid and localized plaque induction, bypassing the prolonged timeline required for spontaneous atherosclerosis in genetic models[81]. It uniquely integrates hemodynamic stress with metabolic dysregulation and mimics the multifactorial etiology of human atherosclerosis.
However, this model also has several limitations. Plaque formation primarily results from mechanical injury and hyperlipidemia, a pathogenic mechanism that differs fundamentally from the natural progression of human atherosclerosis driven by chronic endothelial dysfunction[82]. Furthermore, lesions predominantly exhibit neointimal hyperplasia rather than complex advanced plaques with necrotic cores[83].
This model possesses unique utility for simulating hemodynamics. Therefore, it is a critical research tool to elucidate atherosclerosis biomechanical mechanisms and identify therapeutic interventions targeting atherosclerosis induced by disturbed blood flow[84,85].
Ldlr-ASO mice
The Ldlr-ASO mouse model employs chemically modified ASOs to transiently suppress hepatic Ldlr expression via intraperitoneal administration[86,87]. This pharmacological approach mimics acquired LDLR dysfunction without requiring genetic modifications. In a typical protocol, 10-week-old male or female C57Bl6/J mice are injected weekly with Ldlr-ASO (5 mg/kg) for 16 weeks[88-90]. The treatment thus reduces hepatic Ldlr mRNA and protein expression to levels comparable with those in Ldlr-/- mice[75]. When combined with HFD, Ldlr-ASO-treated mice exhibit severe hypercholesterolemia and accelerated atherosclerosis within nine weeks[88]. Lesions predominantly localize to the aortic root and brachiocephalic artery, displaying key pathological features such as lipid-rich cores, macrophage infiltration, and calcification[88].
The Ldlr-ASO model offers several advantages. Its temporal control over LDLR suppression enables studies on plaque regression or therapeutic reversal of hypercholesterolemia[91]. Additionally, the model mimics acquired LDLR deficiency, making it relevant to patients with LDLR dysfunction due to mutations or metabolic disorders[92]. By utilizing wild-type mice, it also avoids developmental adaptations inherent to lifelong Ldlr knockout strains.
However, there are some limitations in this model. Sustaining LDLR suppression depends on repeated ASO dosing, as exemplified by weekly injections. This requirement complicates experimental workflows and increases variability. Furthermore, the hepatocyte-specific action of ASOs limits investigation into extrahepatic roles of LDLR in atherosclerosis, such as its expression in macrophages or endothelial cells.
The Ldlr-ASO model provides a valuable platform for evaluating anti-atherosclerotic drugs. For instance, it was employed in a study testing empagliflozin—a Food and Drug Administration (FDA)-approved antidiabetic with various cardiorenal benefits—for its effect on atherosclerosis[93]. Empagliflozin-treated mice exhibited reduced atherosclerotic plaque area, along with decreased lipid content and macrophage infiltration, and higher collagen content.
This model's reversibility of atherosclerosis and compatibility with wild-type backgrounds make it particularly suitable for high-throughput screening of potential anti-atherosclerotic pharmacotherapies. This advantage bridges translational gaps between preclinical discovery and clinical application.
AAV8-Pcsk9-D377Y-injected mice
The AAV8-Pcsk9-D377Y mouse model is generated by intravenous injection of an AAV8 vector expressing a gain-of-function mutant human PCSK9 gene (PCSK9-D377Y)[94]. This mutation disrupts the autocatalytic cleavage of PCSK9, stabilizing its interaction with the LDLR and enhancing LDLR degradation in hepatocytes[95]. Following AAV8-mediated hepatic transduction, mice develop persistent hypercholesterolemia due to reduced LDLR recycling. This results in elevated plasma LDL-C levels. When combined with a 12-week HFD, these mice exhibit accelerated atherosclerosis, characterized by macrophage infiltration and fibrous cap formation[96].
This model bypasses the need for germline genetic modifications and its stable transgene expression further supports long-term investigations[97]. Nevertheless, this model has several limitations. Sex differences in atherosclerosis susceptibility persist across specific genetic backgrounds[98]. Similar to most current models, this model fails to recapitulate spontaneous plaque rupture. Furthermore, inter-individual variability in atherosclerotic lesion progression and drug response has been observed in mice treated with AAV8-Pcsk9-D377Y[99].
The AAV8-Pcsk9-D377Y injection model offers a powerful tool for atherosclerosis research. For example, it is used to reveal that serine/threonine protein kinase 25 is a key pro-atherogenic driver[100]. In addition, it has also been applied to elucidate the role of scavenger receptor class B type 1 in vascular endothelium in promoting LDL transcytosis and accumulation[101]. Thus, this model is effective for identifying and characterizing pivotal regulators of atherogenesis.
Western diet (WD)-fed SR-BI∆CT/∆CT/Ldlr-/- mice
The western diet (WD)-fed SR-BI∆CT/∆CT/Ldlr-/- mouse model features combined defects in the carboxyl-terminus of scavenger receptor class B type I and LDLR deletion[73]. This model provides a novel system for studying atherosclerosis pathogenesis. The SR-BI mutation disrupts the cholesterol transport function of SR-BI, impairing high-density lipoprotein (HDL)-mediated reverse cholesterol transport[102]. Concomitantly, the Ldlr-/- mutation abolishes LDL clearance dependent on LDL receptors. Consequently, WD-fed mice develop severe advanced atherosclerosis in the coronary, brachiocephalic, and carotid arteries[73]. This pathology culminates in spontaneous plaque rupture, myocardial infarction, and stroke. Following 26 weeks of WD feeding, this model exhibits high mortality due to these major adverse cardiovascular events. Supporting evidence includes significantly elevated circulating high-sensitivity cardiac troponin I and enhanced neutrophil extracellular trap formation within lesions compared to controls[73].
While this model effectively mimics advanced plaque instability and clinical sequelae, a key limitation is the extended 26-week modeling time[73]. Nevertheless, the SR-BI∆CT/∆CT/Ldlr-/- model robustly mimics late-stage atherosclerosis and its catastrophic complications. Consequently, this model provides a valuable preclinical tool for evaluating therapeutics targeting the prevention of major adverse cardiac events.
CURRENT LIMITATIONS OF MOUSE MODELS IN TRANSLATIONAL APPLICATIONS
Mouse atherosclerosis models face important translational limitations due to metabolic and pathological disparities and oversimplified disease complexity. Mice predominantly transport cholesterol via HDL and exhibit rapid biliary cholesterol clearance[103]. This contrasts with human LDL dominance, leading to divergent drug responses across species[104,105]. Pathologically, murine atherosclerotic plaques develop primarily in the aortic root and aorta, with rare occurrence in human-susceptible coronary arteries[13,106]. Additionally, murine plaques lack critical human coronary features such as thin fibrous caps and spontaneous rupture. Therefore, artificial interventions—typically surgical stenosis induction—are required to model thrombosis in these models[107,108]. Inadequate modeling of neutrophil-driven inflammation and insufficient integration of comorbidities further constrain clinical relevance[109]. These translational gaps underscore the urgent need for advanced models capable of replicating human lipid networks, plaque dynamics, and systemic disease interactions [Figure 2; Table 2].
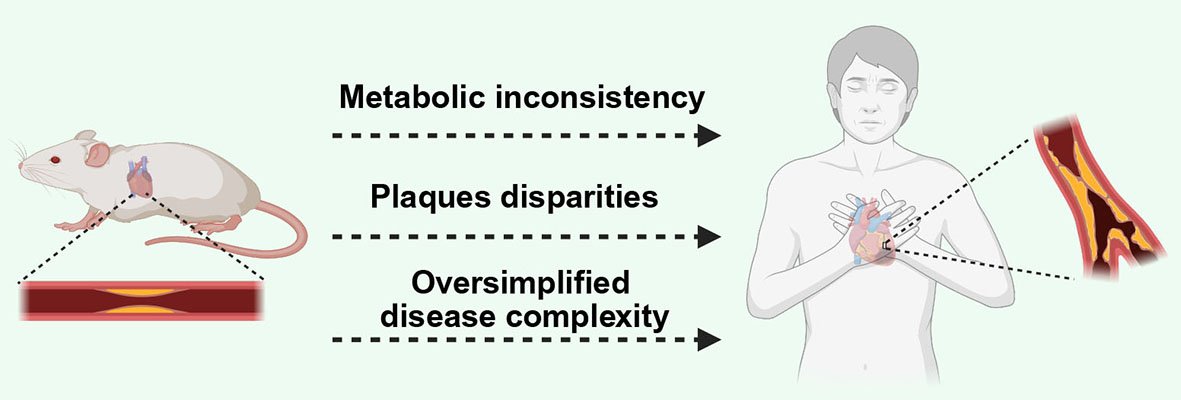
Figure 2. The difference of atherosclerosis between human and mouse. Divergences between human and murine atherosclerosis primarily manifest as: metabolic profiles, plaque phenotypes, and oversimplification of disease complexity. Created in BioRender. kaisi, Y. (2025) https://BioRender.com/n6ysic2.
Key comparative indicators of atherosclerosis between humans and wild-type mice
| Key indicators | Human | Wild-type mice | References |
| Major circulating lipoprotein | ApoB-containing lipoproteins (primarily LDL) | ApoA-I-containing lipoproteins (primarily HDL) | [110,111] |
| LDL/HDL ratio | 1.6-4 | 0.1-0.3 | [104,105] |
| CETP activity | Present | Largely absent | [112] |
| Primary plaque location | Coronary arteries, carotid, and cerebral arteries | Aortic root and aorta arch | [13,106] |
| Plaque composition | Complex, large necrotic core, thin fibrous cap, intraplaque hemorrhage | More stable, smaller lipid core, thicker fibrous cap | [116,117] |
| Plaque rupture and complications | Common (MI, stroke) | Extremely rare (requires complex triggering) | [116,118] |
Metabolic and pathological disparities
A critical consideration in the use of murine models for atherosclerosis research is the fundamental difference in lipid metabolism between mice and humans, which poses significant translational challenges. Unlike in humans, where LDL serves as the primary cholesterol carrier and a key atherogenic factor, mice predominantly transport cholesterol via HDL—a profile generally regarded as atheroprotective[110,111]. This divergence stems from differences in lipid regulatory pathways. Mice lack CETP, which in humans facilitates the transfer of cholesteryl esters from HDL to LDL and VLDL[112]. As a result, wild-type mice exhibit a plasma lipid profile characterized by high HDL and low LDL levels, a pattern associated with low cardiovascular risk in humans. This inherent metabolic distinction necessitates the use of genetically engineered mouse strains to mimic human-like dyslipidemia. It also fundamentally shapes the pathophysiology of lesion development in these models.
Mouse models of atherosclerosis exhibit inherent metabolic and pathological disparities that reduce their translational accuracy to human disease, despite their widespread use[103,113]. Metabolically, mice exhibit fundamental differences from humans in lipid homeostasis, particularly through predominant HDL transport of cholesterol versus human LDL dominance[114]. Moreover, their efficient biliary system rapidly converts cholesterol to bile acids—a process substantially attenuated in humans[114]. These differences are exemplified by the failure of CETP inhibitors. While these inhibitors elevate HDL and reduce LDL in mice, they show limited clinical benefits in human trials[115]. Such metabolic divergence complicates the extrapolation of therapeutic outcomes.
Pathologically, murine atherosclerotic plaques differ from human lesions in location and composition. Advanced human plaques predominantly develop in coronary arteries, exhibiting high-risk features such as thin fibrous caps, large necrotic cores, and intraplaque hemorrhage[116]. In contrast, mouse lesions primarily form in the aortic root and arch, demonstrating stable fibrous caps with minimal calcification or thrombotic complications[13,117]. For instance, widely used models such as ApoE-/- or Ldlr-/- mice rarely develop spontaneous plaque rupture, necessitating invasive interventions to mimic clinical events[118]. Murine immune responses are different from human inflammatory profiles, particularly regarding neutrophil and monocyte subset dominance during early atherogenesis[119]. Consequently, these immunological differences limit the translational relevance of immunomodulatory therapies, underscoring the challenges in modeling the progression and complications of human atherosclerosis.
Inadequate replication of disease complexity
Current mouse models often fail to capture the multifactorial complexity of human atherosclerosis, particularly its interplay with comorbid conditions and environmental factors. Human atherosclerosis frequently coexists with metabolic syndrome, chronic kidney disease, or autoimmune disorders. These comorbidities synergistically accelerate plaque progression and complications[120-122]. However, most mouse models isolate atherosclerosis from these comorbidities, limiting their translational relevance. For instance, while ApoE-/- mice fed a HFD develop hyperlipidemia and plaques, they lack the insulin resistance, adipose tissue inflammation, or gut microbiota dysbiosis characteristic of human metabolic syndrome[123,124]. These models are also combined with streptozotocin-induced diabetes or angiotensin II infusion (to model hypertension), yet they fail to fully replicate human atherosclerotic disease[125,126]. In summary, current modeling methods overlook the systemic crosstalk between metabolic organs and vascular systems.
Furthermore, mouse models inadequately simulate the characteristics of advanced human atherosclerosis, such as plaque rupture, erosion, and atherothrombosis[127]. Invasive techniques such as tandem stenosis can induce plaque instability[128]. However, these approaches introduce non-physiological mechanical/chemical stressors absent in natural disease progression. For example, PCL in ApoE-/- mice causes flow-induced endothelial damage but fails to replicate the spontaneous rupture of thin-cap fibroatheromas seen in humans[128]. Consequently, some therapies such as PCSK9 inhibitors and CETP inhibitors showing efficacy in mouse models often fail in clinical trials[129-131]. These translational failures underscore the urgent need for disease models that authentically mimic human pathological complexity.
CONCLUSION
Mouse models have been indispensable in unraveling the pathophysiology of atherosclerosis and advancing therapeutic innovation. Classic models, such as ApoE-/- and Ldlr-/- mice, have been used to study lipid metabolism, inflammation, and plaque dynamics. Other genetically modified strains alongside novel mouse models have enhanced translational relevance by replicating human-like dyslipidemia and complex disease phenotypes[9,10]. These models have been used for preclinical validation of lipid-lowering agents, anti-inflammatory therapies, and anti-atherosclerotic drugs, bridging mechanistic discoveries to clinical applications[109,127]. However, persistent metabolic disparities and incomplete replication of human plaque complexity remain key limitations[103,132]. These unresolved issues highlight the necessity of continuous improvement of the mouse atherosclerosis model.
In this context, progeria mouse models, such as those carrying mutations in the lamin A/C (LMNA) gene (e.g., LmnaG609G/G609G), offer a unique perspective by exhibiting accelerated cardiovascular aging[133]. These models develop extensive atherosclerosis and vascular calcification at an early age, alongside characteristic arterial stiffening and endothelial dysfunction[134]. The pronounced and rapid progression of cardiovascular pathology in these mice provides a valuable system for studying the interplay between premature aging, metabolic stress, and atherogenesis. It thereby complements insights gained from classic hyperlipidemic models.
Atherosclerosis research has long relied on classic hypercholesterolemic mouse models, such as the ApoE-/- and Ldlr-/- strains, typically maintained on a HFD to drive plaque development. Given that metabolic syndrome represents a major risk factor for atherosclerosis, more advanced models capable of recapitulating its complex pathophysiology are required[135]. To address this, several innovative models have emerged. For example, genetic and viral tools—including Ldlr-ASO and AAV8-Pcsk9-D377Y injection—allow precise temporal and combinatorial control over severe hypercholesterolemia[75,95]. Notably, the WD-fed SR-BI∆CT/∆CT/Ldlr-/- mouse model exhibits human-like atherogenic dyslipidemia and forms unstable plaques, establishing it as a key model for studying metabolic syndrome–accelerated atherosclerosis[73]. This progression reflects a shift toward models that integrate dyslipidemia, insulin resistance, and plaque instability, thereby enhancing the translational relevance of preclinical research for cardiovascular drug discovery.
Future efforts should prioritize developing next-generation models that integrate multifactorial disease drivers. For example, engineering mouse models involves integrating humanized lipoprotein profiles with comorbidities, including diabetes and hypertension. These can better mimic the disease interaction underlying clinical atherosclerosis. Additionally, immune-metabolic perturbations may replicate human plaque vulnerability and spontaneous thrombosis. These innovations can be synergized with high-resolution phenotyping tools, particularly intravital imaging and single-cell omics. This integration can accurately dissect the progression and therapeutic response of atherosclerosis.
Beyond their role in drug validation for translational applications, these atherosclerosis models are equally pivotal for mechanistic investigation and drug screening. Classical genetic knockout models, such as Ldlr-/- and Apoe-/- mice, remain foundational for dissecting gene functions in atherogenesis due to their robust phenotypes[32,56]. For studying specific mechanistic aspects, such as localized hemodynamic stress or HDL metabolism defects, intervention-based models are often employed. These approaches, including PCL combined with a HFD or the WD-fed SR-BI∆CT/∆CT/Ldlr-/- mouse, can offer more precise mechanistic insights[73,80]. For drug screening and preclinical validation, models that allow rapid, controllable, and human-relevant disease induction are generally favored. The Ldlr-ASO mice and AAV8-Pcsk9-D377Y injection models exemplify this approach by rapidly establishing severe hypercholesterolemia and atherosclerosis within weeks[88,96]. Furthermore, the Ldlr-ASO model enables reversible pharmacological suppression of LDLR, making it suitable for studying the effects of gene expression restoration. Therefore, the choice of experimental model should be guided by the specific research objective. Classical knockout models such as Apoe-/- and Ldlr-/- mice are suitable for long-term pathogenesis studies. Intervention-based models such as ligation or dual-knockout strains target specific mechanistic pathways, whereas ASO- and adeno-associated virus (AAV)-mediated systems offer efficient, flexible platforms for high-throughput therapeutic screening.
In addition to selecting models based on research objectives, translational success also depends on adopting emerging technologies and interdisciplinary approaches. Humanized mouse models reconstituted with patient-derived cells such as induced pluripotent stem cells (iPSCs)-derived macrophages or human microbiota could enable personalized therapeutic testing[136]. Artificial intelligence-driven integration of multi-omics data from murine and human cohorts may identify evolutionarily conserved pathways for targeted intervention[137]. Furthermore, fostering collaboration between preclinical researchers, clinicians, and bioengineers will accelerate the translation of discoveries into clinical applications. By addressing existing limitations, next-generation atherosclerosis models will be helpful to clarify unresolved pathological mechanisms and open up new areas to combat this global disease.
DECLARATIONS
Acknowledgments
We thank kaisi, Y. for assistance with figures creation using BioRender.com. Graphic abstract was created in BioRender. kaisi, Y. (2025) https://BioRender.com/tyhvysg.
Authors' contributions
Wrote the original manuscript: Yang Z
Critically reviewed the manuscript: She ZG
Both authors contributed to the article and approved the submitted version.
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
Not applicable.
Financial support and sponsorship
This work was supported by grants from the National Natural Science Foundation of China (No. 882270390).
Conflict of interest
Both authors declared that there are no conflicts of interest.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Martin SS, Aday AW, Allen NB, et al. 2025 heart disease and stroke statistics: a report of US and global data from the American Heart Association. Circulation. 2025;151:e41-660.
2. Jinson S, Zhang Z, Lancaster GI, Murphy AJ, Morgan PK. Iron, lipid peroxidation, and ferroptosis play pathogenic roles in atherosclerosis. Cardiovasc Res. 2025;121:44-61.
3. Gusdorf J, Faridi KF, Wang TY, et al. Inpatient outcomes for patients with peripheral artery disease hospitalized for acute myocardial infarction. J Am Heart Assoc. 2025;14:e040526.
4. Bookmeyer CHM, Correig FX, Masana L, Magni P, Yanes Ó, Vinaixa M. Advancing atherosclerosis research: the power of lipid imaging with MALDI-MSI. Atherosclerosis. 2025;403:119130.
5. Schaefer EJ, Asztalos BF, Vaisar T, et al. High density lipoprotein particle composition, functionality, deficiency, and atherosclerotic cardiovascular disease risk: a review. Curr Atheroscler Rep. 2025;27:62.
6. Botros M, Fadah K, Mukherjee D. The role of inflammatory response in the development of atherosclerosis, myocardial infarction, and remodeling. Vessel Plus. 2024;8:31.
7. Li H, Liu T, Shi X, et al. Mechanisms and therapeutic potential of pharmacological agents targeting inflammasomes. Biomed Pharmacother. 2025;189:118164.
8. Kraler S, Mueller C, Libby P, Bhatt DL. Acute coronary syndromes: mechanisms, challenges, and new opportunities. Eur Heart J. 2025;46:2866-89.
9. Ilyas I, Little PJ, Liu Z, et al. Mouse models of atherosclerosis in translational research. Trends Pharmacol Sci. 2022;43:920-39.
10. Jiang H, Liao Y, Zhu M, et al. Innovative atherosclerosis models: advancing pathophysiology and translational research. Research. 2025;8:0617.
11. Strijdhorst A, Vos WG, Bosmans LA, et al. Accelerated atherosclerosis associated with immune checkpoint inhibitors: a systematic review and meta-analysis of pre-clinical studies. Atherosclerosis. 2025;405:119219.
12. Wang R, Zhang H, Li S, et al. Current progress of in vitro vascular models on microfluidic chips. Biofabrication. 2025;17:022004.
13. Zhao Y, Qu H, Wang Y, Xiao W, Zhang Y, Shi D. Small rodent models of atherosclerosis. Biomed Pharmacother. 2020;129:110426.
14. Poznyak AV, Silaeva YY, Orekhov AN, Deykin AV. Animal models of human atherosclerosis: current progress. Braz J Med Biol Res. 2020;53:e9557.
15. Heo KS, Phan LP, Le NTT, Jin Y. Mechanistic insights and emerging therapeutic strategies targeting endothelial dysfunction in cardiovascular diseases. Arch Pharmacal Res. 2025;48:305-32.
16. Xu S, Ilyas I, Little PJ, et al. Endothelial dysfunction in atherosclerotic cardiovascular diseases and beyond: from mechanism to pharmacotherapies. Pharmacol Rev. 2021;73:924-67.
17. Oskroba A, Bartusik-Aebisher D, Myśliwiec A, et al. Photodynamic therapy and cardiovascular diseases. Int J Mol Sci. 2024;25:2974.
18. Mormone A, Tortorella G, Esposito F, et al. Advances in pharmacological approaches for managing hypercholesterolemia: a comprehensive overview of novel treatments. Biomedicines. 2024;12:432.
19. La Chica Lhoëst MT, Martinez A, Claudi L, et al. Mechanisms modulating foam cell formation in the arterial intima: exploring new therapeutic opportunities in atherosclerosis. Front Cardiovasc Med. 2024;11:1381520.
20. Carrizzo A, Izzo C, Forte M, et al. A novel promising frontier for human health: the beneficial effects of nutraceuticals in cardiovascular diseases. Int J Mol Sci. 2020;21:8706.
21. Wang X, Xie Z, Zhang J, et al. Interaction between lipid metabolism and macrophage polarization in atherosclerosis. iScience. 2025;28:112168.
22. Iliodromitis K, Seyfarth M, Balogh Z, Bogossian H, Iliodromitis E, Triposkiadis F. Anti-inflammatory interventions in coronary artery disease: antipodal responses requiring targeted therapeutic strategies. Basic Res Cardiol. 2025;120:597-618.
23. Chen R, Zhou Z, Song Z, et al. Tangzhiqing exacerbates oxidized low-density lipoprotein-induced cell pyroptosis through activation of NLRP3 inflammasome in human umbilical vein endothelial cells. Cell Biol Int. 2025;49:1173-83.
24. Lin L, Deng KQ, Chen Z, et al. Lipoprotein(a) distribution and its association with carotid arteriopathy in the Chinese population. Atherosclerosis. 2023;372:1-9.
25. Wang Z, Ye M, Zhang XJ, et al. Impact of NAFLD and its pharmacotherapy on lipid profile and CVD. Atherosclerosis. 2022;355:30-44.
26. Kwon H, Ryu JC, Cha JK, et al. Low-density lipoprotein cholesterol level, the lower the better? Analysis of Korean patients in the treat stroke to target trial. J Stroke. 2025;27:228-36.
27. Tian Y, Zong Y, Pang Y, et al. Platelets and diseases: signal transduction and advances in targeted therapy. Signal Transduct Target Ther. 2025;10:159.
28. Rauch PJ, Gopakumar J, Silver AJ, et al. Loss-of-function mutations in Dnmt3a and Tet2 lead to accelerated atherosclerosis and concordant macrophage phenotypes. Nat Cardiovasc Res. 2023;2:805-18.
29. Getz GS, Reardon CA. Do the Apoe-/- and Ldlr-/- mice yield the same insight on atherogenesis? Arterioscler Thromb Vasc Biol. 2016;36:1734-41.
30. Emini Veseli B, Perrotta P, De Meyer GRA, et al. Animal models of atherosclerosis. Eur J Pharmacol. 2017;816:3-13.
31. Zhang Y, Ren D, Liu Y, et al. Mechanism of red yeast rice in the improvement of atherosclerosis in apolipoprotein E-deficient mice explored through metabolomics combined with serum pharmacochemistry and network pharmacology. Plant Foods Hum Nutr. 2025;80:140.
32. Rosenfeld ME, Polinsky P, Virmani R, Kauser K, Rubanyi G, Schwartz SM. Advanced atherosclerotic lesions in the innominate artery of the ApoE knockout mouse. Arterioscler Thromb Vasc Biol. 2000;20:2587-92.
33. van Leeuwen M, Gijbels MJ, Duijvestijn A, et al. Accumulation of myeloperoxidase-positive neutrophils in atherosclerotic lesions in LDLR-/- mice. Arterioscler Thromb Vasc Biol. 2008;28:84-9.
34. Getz GS, Reardon CA. PCSK9 and lipid metabolism and atherosclerosis: animal models. Vessel Plus. 2021;5:17.
35. Meyrelles SS, Peotta VA, Pereira TM, Vasquez EC. Endothelial dysfunction in the apolipoprotein E-deficient mouse: insights into the influence of diet, gender and aging. Lipids Health Dis. 2011;10:211.
36. Nieswandt B, Aktas B, Moers A, Sachs UJ. Platelets in atherothrombosis: lessons from mouse models. J Thromb Haemost. 2005;3:1725-36.
37. Inia JA, van Nieuwkoop-van Straalen A, Jukema JW, et al. Efficacy of a novel PCSK9 inhibitory peptide alone and with evinacumab in a mouse model of atherosclerosis. J Lipid Res. 2025;66:100753.
38. Ruotsalainen AK, Lappalainen JP, Heiskanen E, et al. Nuclear factor E2-related factor 2 deficiency impairs atherosclerotic lesion development but promotes features of plaque instability in hypercholesterolaemic mice. Cardiovasc Res. 2019;115:243-54.
39. Piedrahita JA, Zhang SH, Hagaman JR, Oliver PM, Maeda N. Generation of mice carrying a mutant apolipoprotein E gene inactivated by gene targeting in embryonic stem cells. Proc Natl Acad Sci USA. 1992;89:4471-5.
40. Liu D, Mai D, Jahn AN, et al. APOE protects against severe infection with Mycobacterium tuberculosis by restraining production of neutrophil extracellular traps. PLoS Pathog. 2025;21:e1013267.
41. Murphy AJ, Akhtari M, Tolani S, et al. ApoE regulates hematopoietic stem cell proliferation, monocytosis, and monocyte accumulation in atherosclerotic lesions in mice. J Clin Invest. 2011;121:4138-49.
42. Johnson J, Carson K, Williams H, et al. Plaque rupture after short periods of fat feeding in the apolipoprotein E-knockout mouse: model characterization and effects of pravastatin treatment. Circulation. 2005;111:1422-30.
43. Oppi S, Lüscher TF, Stein S. Mouse models for atherosclerosis research-which is my line? Front Cardiovasc Med. 2019;6:46.
44. Fernandez DM, Giannarelli C. Immune cell profiling in atherosclerosis: role in research and precision medicine. Nat Rev Cardiol. 2022;19:43-58.
45. Schroeter MR, Humboldt T, Schäfer K, Konstantinides S. Rosuvastatin reduces atherosclerotic lesions and promotes progenitor cell mobilisation and recruitment in apolipoprotein E knockout mice. Atherosclerosis. 2009;205:63-73.
46. Peng C, Li J, Chen Y, et al. PCSK9 aggravated carotid artery stenosis in ApoE-/- mice by promoting the expression of tissue factors in endothelial cells via the TLR4/NF-κB pathway. Biochem Pharmacol. 2024;225:116314.
47. Guo X, Wang L, Xia X, Wang P, Li X. Effects of atorvastatin and/or probucol on recovery of atherosclerosis in high-fat-diet-fed apolipoprotein E-deficient mice. Biomed Pharmacother. 2019;109:1445-53.
48. Di Marco E, Gray SP, Chew P, et al. Pharmacological inhibition of NOX reduces atherosclerotic lesions, vascular ROS and immune-inflammatory responses in diabetic Apoe-/- mice. Diabetologia. 2014;57:633-42.
49. Jongejan YK, Eikenboom JCJ, Gijbels MJJ, Berbée JFP, van Vlijmen BJM. Atherothrombosis model by silencing of protein C in APOE*3-Leiden.CETP transgenic mice. J Thromb Thrombolysis. 2021;52:715-9.
50. Westerterp M, van der Hoogt CC, de Haan W, et al. Cholesteryl ester transfer protein decreases high-density lipoprotein and severely aggravates atherosclerosis in APOE*3-Leiden mice. Arterioscler Thromb Vasc Biol. 2006;26:2552-9.
51. Paalvast Y, Zhou E, Rozendaal YJW, et al. A systems analysis of phenotype heterogeneity in APOE*3Leiden.CETP mice induced by long-term high-fat high-cholesterol diet feeding. Nutrients. 2022;14:4936.
52. Paalvast Y, Gerding A, Wang Y, et al. Male apoE*3-Leiden.CETP mice on high-fat high-cholesterol diet exhibit a biphasic dyslipidemic response, mimicking the changes in plasma lipids observed through life in men. Physiol Rep. 2017;5:e13376.
53. Kong YY, Li GQ, Zhang WJ, et al. Nicotinamide phosphoribosyltransferase aggravates inflammation and promotes atherosclerosis in ApoE knockout mice. Acta Pharmacol Sin. 2019;40:1184-92.
54. Ishibashi S, Goldstein JL, Brown MS, Herz J, Burns DK. Massive xanthomatosis and atherosclerosis in cholesterol-fed low density lipoprotein receptor-negative mice. J Clin Invest. 1994;93:1885-93.
55. Schmidt HM, Jarrett KE, de Aguiar Vallim TQ, Tarling EJ. Pathways and molecular mechanisms governing LDL receptor regulation. Circ Res. 2025;136:902-19.
56. Hartvigsen K, Binder CJ, Hansen LF, et al. A diet-induced hypercholesterolemic murine model to study atherogenesis without obesity and metabolic syndrome. Arterioscler Thromb Vasc Biol. 2007;27:878-85.
57. Hoekstra M, Van Eck M. HDL is redundant for adrenal steroidogenesis in LDLR knockout mice with a human-like lipoprotein profile. J Lipid Res. 2016;57:631-7.
58. Burke AC, Telford DE, Huff MW. Bempedoic acid: effects on lipoprotein metabolism and atherosclerosis. Curr Opin Lipidol. 2019;30:1-9.
59. Wouters K, Shiri-sverdlov R, Van Gorp PJ, Van Bilsen M, Hofker MH. Understanding hyperlipidemia and atherosclerosis: lessons from genetically modified apoe and ldlr mice. Clin Chem Lab Med. 2005;43:470-9.
60. Ibrahim S, Hartgers ML, Reeskamp LF, et al. LDLR variant classification for improved cardiovascular risk prediction in familial hypercholesterolemia. Atherosclerosis. 2024;397:117610.
61. Stankov S, Cuchel M. Gene editing for dyslipidemias: new tools to “cut” lipids. Atherosclerosis. 2023;368:14-24.
62. Véniant MM, Zlot CH, Walzem RL, et al. Lipoprotein clearance mechanisms in LDL receptor-deficient "Apo-B48-only" and "Apo-B100-only" mice. J Clin Investig. 1998;102:1559-68.
63. Calara F, Silvestre M, Casanada F, Yuan N, Napoli C, Palinski W. Spontaneous plaque rupture and secondary thrombosis in apolipoprotein E-deficient and LDL receptor-deficient mice. J. Pathol. 2001;195:257-63.
64. Hellberg S, Sippola S, Liljenbäck H, et al. Effects of atorvastatin and diet interventions on atherosclerotic plaque inflammation and [18F]FDG uptake in Ldlr-/-Apob mice. Atherosclerosis. 2017;263:369-76.
65. Bhakta S, Kodama H, Mimaki M, Tsukahara T. Restoration of genetic code in macular mouse fibroblasts via APOBEC1-mediated RNA editing. Biomolecules. 2025;15:136.
66. Miyajima C, Iwaki T, Umemura K, Ploplis VA, Castellino FJ. Characterization of atherosclerosis formation in a murine model of type IIa human familial hypercholesterolemia. Biomed Res Int. 2018;2018:1878964.
67. Dutta R, Singh U, Li T, Fornage M, Teng B. Hepatic gene expression profiling reveals perturbed calcium signaling in a mouse model lacking both LDL receptor and Apobec1 genes. Atherosclerosis. 2003;169:51-62.
68. Shao B, Shimizu-albergine M, Kramer F, et al. A targeted proteomics method for quantifying plasma apolipoprotein kinetics in individual mice using stable isotope labeling. J Lipid Res. 2024;65:100531.
69. Basu D, Bornfeldt KE. Hypertriglyceridemia and atherosclerosis: using human research to guide mechanistic studies in animal models. Front Endocrinol. 2020;11:504.
70. Hajighasemi S, Mahdavi Gorabi A, Bianconi V, et al. A review of gene- and cell-based therapies for familial hypercholesterolemia. Pharmacol Res. 2019;143:119-32.
71. Getz GS, Reardon CA. Insights from murine studies on the site specificity of atherosclerosis. Int J Mol Sci. 2024;25:6375.
72. Ajoolabady A, Pratico D, Mazidi M, et al. PCSK9 in metabolism and diseases. Metabolism. 2025;163:156064.
73. Shamsuzzaman S, Deaton RA, Salamon A, et al. Novel mouse model of myocardial infarction, plaque rupture, and stroke shows improved survival with myeloperoxidase inhibition. Circulation. 2024;150:687-705.
74. Liang Y, Liu J, Zhang C, et al. Integrating serum pharmacochemistry and metabolomics to reveal the potential effective ingredients and mechanism of Huangqi Chifeng Tang intervening carotid atherosclerosis. J Ethnopharmacol. 2025;351:120065.
75. Gomes D, Wang S, Goodspeed L, et al. Comparison between genetic and pharmaceutical disruption of Ldlr expression for the development of atherosclerosis. J Lipid Res. 2022;63:100174.
76. Bjørklund MM, Hollensen AK, Hagensen MK, et al. Induction of atherosclerosis in mice and hamsters without germline genetic engineering. Circ Res. 2014;114:1684-9.
77. Park C, Baek KI, Hung R, et al. Disturbed flow induces reprogramming of endothelial cells to immune-like and foam cells under hypercholesterolaemia during atherogenesis. Cardiovasc Res. 2025;121:2679-99.
78. Zhou X, Kuenne C, Günther S, et al. Nuclear eNOS interacts with and S-nitrosates ADAR1 to modulate type I interferon signaling and endothelial function. Circulation. 2025;152:1781-99.
79. Jiang D, Liu H, Zhu G, et al. Endothelial PHACTR1 promotes endothelial activation and atherosclerosis by repressing PPARγ activity under disturbed flow in mice. Arterioscler Thromb Vasc Biol. 2023;43:8.
80. Mitra R, Qiao J, Madhavan S, et al. The comparative effects of high fat diet or disturbed blood flow on glycocalyx integrity and vascular inflammation. Transl Med Commun. 2018;3:10.
81. Nam D, Ni C, Rezvan A, et al. Partial carotid ligation is a model of acutely induced disturbed flow, leading to rapid endothelial dysfunction and atherosclerosis. Am J Physiol Heart Circ Physiol. 2009;297:H1535-43.
82. Dib L, Koneva LA, Edsfeldt A, et al. Lipid-associated macrophages transition to an inflammatory state in human atherosclerosis, increasing the risk of cerebrovascular complications. Nat Cardiovasc Res. 2023;2:656-72.
83. Cho M, Hwang JS, Kim KR, Kim JK. Wall shear stress (WSS) analysis in atherosclerosis in partial ligated apolipoprotein E knockout mouse model through computational fluid dynamics (CFD). Int J Mol Sci. 2024;25:9877.
84. Chien C, Li JY, Chien Y, et al. METTL3-dependent N6-methyladenosine RNA modification mediates the atherogenic inflammatory cascades in vascular endothelium. Proc Natl Acad Sci USA. 2021;118:e2025070118.
85. Hu S, Liu Y, You T, et al. Vascular semaphorin 7A upregulation by disturbed flow promotes atherosclerosis through endothelial β1 integrin. Arterioscler Thromb Vasc Biol. 2018;38:335-43.
86. Pinos I, Coronel J, Albakri A, et al. β-carotene accelerates the resolution of atherosclerosis in mice. eLife. 2024;12:RP87430.
87. Chen Y, Johnson SM, Burr SD, et al. Absence of the intracellular lipolytic inhibitor G0S2 enhances intravascular triglyceride clearance and abolishes diet-induced hypertriglyceridemia. J Clin Investig. 2025;135:e181754.
88. Basu D, Hu Y, Huggins L, et al. Novel reversible model of atherosclerosis and regression using oligonucleotide regulation of the LDL receptor. Circ Res. 2018;122:560-7.
89. Josefs T, Basu D, Vaisar T, et al. Atherosclerosis regression and cholesterol efflux in hypertriglyceridemic mice. Circ Res. 2021;128:690-705.
90. Bashore AC, Liu M, Key CC, et al. Targeted deletion of hepatocyte Abca1 increases plasma HDL (high-density lipoprotein) reverse cholesterol transport via the LDL (low-density lipoprotein) receptor. Arterioscler Thromb Vasc Biol. 2019;39:1747-61.
91. Burr SD, Chen Y, Hartley CP, Zhao X, Liu J. Replacement of saturated fatty acids with linoleic acid in western diet attenuates atherosclerosis in a mouse model with inducible ablation of hepatic LDL receptor. Sci Rep. 2023;13:16832.
92. Mullick AE, Fu W, Graham MJ, et al. Antisense oligonucleotide reduction of apoB-ameliorated atherosclerosis in LDL receptor-deficient mice. J Lipid Res. 2011;52:885-96.
93. Pennig J, Scherrer P, Gissler MC, et al. Glucose lowering by SGLT2-inhibitor empagliflozin accelerates atherosclerosis regression in hyperglycemic STZ-diabetic mice. Sci Rep. 2019;9:17937.
94. Ansari A, Yadav PK, Zhou L, et al. Casz1 and Znf101/Zfp961 differentially regulate apolipoproteins A1 and B, alter plasma lipoproteins, and reduce atherosclerosis. JCI Insight. 2025;10:e182260.
95. Yurtseven E, Ural D, Baysal K, Tokgözoğlu L. An update on the role of PCSK9 in atherosclerosis. J Atheroscler Thromb. 2020;27:909-18.
96. Roche-molina M, Sanz-rosa D, Cruz FM, et al. Induction of sustained hypercholesterolemia by single adeno-associated virus-mediated gene transfer of mutant hPCSK9. Arterioscler Thromb Vasc Biol. 2015;35:50-9.
97. Caruana V, Giles BH, Kukolj N, Juran R, Baglole CJ, Mann KK. Chronic exposure to E-cigarette aerosols potentiates atherosclerosis in a sex-dependent manner. Toxicol Appl Pharmacol. 2024;492:117095.
98. Buchmann GK, Schürmann C, Warwick T, et al. Deletion of NoxO1 limits atherosclerosis development in female mice. Redox Biol. 2020;37:101713.
99. Jia M, Li Q, Guo J, et al. Deletion of BACH1 attenuates atherosclerosis by reducing endothelial inflammation. Circ Res. 2022;130:1038-55.
100. Cansby E, Magnusson E, Nuñez-Durán E, et al. STK25 regulates cardiovascular disease progression in a mouse model of hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2018;38:1723-37.
101. Huang L, Chambliss KL, Gao X, et al. SR-B1 drives endothelial cell LDL transcytosis via DOCK4 to promote atherosclerosis. Nature. 2019;569:565-9.
102. Acton S, Rigotti A, Landschulz KT, Xu S, Hobbs HH, Krieger M. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science. 1996;271:518-20.
103. Kaabia Z, Poirier J, Moughaizel M, et al. Plasma lipidomic analysis reveals strong similarities between lipid fingerprints in human, hamster and mouse compared to other animal species. Sci Rep. 2018;8:15893.
104. Ellis ECS, Nauglers S, Parini P, et al. Mice with chimeric livers are an improved model for human lipoprotein metabolism. PLoS ONE. 2013;8:e78550.
105. Sun T, Chen M, Shen H, et al. Predictive value of LDL/HDL ratio in coronary atherosclerotic heart disease. BMC Cardiovasc Disord. 2022;22:273.
106. Sari G, Meester EJ, Van Der Zee LC, et al. A mouse model of humanized liver shows a human-like lipid profile, but does not form atherosclerotic plaque after western type diet. Biochem Biophys Res Commun. 2020;524:510-5.
107. Schwartz SM, Galis ZS, Rosenfeld ME, Falk E. Plaque rupture in humans and mice. Arterioscler Thromb Vasc Biol. 2007;27:705-13.
108. Marino A, Zhang Y, Rubinelli L, Riemma MA, Ip JE, Di Lorenzo A. Pressure overload leads to coronary plaque formation, progression, and myocardial events in ApoE-/- mice. JCI Insight. 2019;4:e128220.
109. Gisterå A, Ketelhuth DF, Malin SG, Hansson GK. Animal models of atherosclerosis-supportive notes and tricks of the trade. Circ Res. 2022;130:1869-87.
110. Wan M, Pan S, Shan B, et al. Lipid metabolic reprograming: the unsung hero in breast cancer progression and tumor microenvironment. Mol Cancer. 2025;24:61.
111. Checkouri E, Blanchard V, Meilhac O. Macrophages in atherosclerosis, first or second row players? Biomedicines. 2021;9:1214.
112. Ebert ML, Schmidt VF, Pfaff L, Von Thaden A, Kimm MA, Wildgruber M. Animal models of neointimal hyperplasia and restenosis. JACC Basic Transl Sci. 2021;6:900-17.
113. Hansson GK, Heistad DD. Two views on plaque rupture. Arterioscler Thromb Vasc Biol. 2007;27:697.
114. Rosenson RS, Brewer HB, Davidson WS, et al. Cholesterol efflux and atheroprotection: advancing the concept of reverse cholesterol transport. Circulation. 2012;125:1905-19.
115. Schmidt AF, Hunt NB, Gordillo-marañón M, et al. Cholesteryl ester transfer protein (CETP) as a drug target for cardiovascular disease. Nat Commun. 2021;12:5640.
116. Aizaz M, Bierens J, Gijbels MJ, et al. Differentiation of atherosclerotic carotid plaque components with dual-energy computed tomography. Investig Radiol. 2025;60:508-16.
117. Delwarde C, Aikawa M. Novel mouse model of late-stage coronary atherosclerosis with features of plaque rupture and stroke. Circulation. 2024;150:706-9.
118. Golforoush P, Yellon DM, Davidson SM. Mouse models of atherosclerosis and their suitability for the study of myocardial infarction. Basic Res Cardiol. 2020;115:73.
120. Alebna PL, Ambrosio M, Martin M, et al. Association of Lipoprotein(a) with cardiovascular events among individuals with autoimmune conditions. Atherosclerosis. 2025;406:119244.
121. Kunimura A, Amano T, Uetani T, et al. Prognostic impact of concurrence of metabolic syndrome and chronic kidney disease in patients undergoing coronary intervention: involvement of coronary plaque composition. J Cardiol. 2013;61:189-95.
122. Reiss AB, Glass DS, Voloshyna I, Glass AD, Kasselman LJ, De Leon J. Obesity and atherosclerosis: the exosome link. Vessel Plus. 2020;4:19.
123. Von Scheidt M, Zhao Y, Kurt Z, et al. Applications and limitations of mouse models for understanding human atherosclerosis. Cell Metab. 2017;25:248-61.
124. Zhang Y, Cheng Z, Hong L, et al. Apolipoprotein E (ApoE) orchestrates adipose tissue inflammation and metabolic disorders through NLRP3 inflammasome. Mol Biomed. 2023;4:47.
125. Cefalu WT. Animal models of type 2 diabetes: clinical presentation and pathophysiological relevance to the human condition. ILAR J. 2006;47:186-98.
126. Daugherty A, Sawada H, Sheppard MB, Lu HS. Angiotensinogen as a therapeutic target for cardiovascular and metabolic diseases. Arterioscler Thromb Vasc Biol. 2024;44:1021-30.
127. Lee YT, Lin HY, Chan YWF, et al. Mouse models of atherosclerosis: a historical perspective and recent advances. Lipids Health Dis. 2017;16:12.
128. Wang J, Shan S, Lyu A, Wan Y, Zhang J. A novel model of myocardial infarction based on atherosclerosis in mice. Biochem Biophys Res Commun. 2021;576:100-7.
129. Bimal T, Ayyalu T, Safarova MS, et al. Inadequate response to PCSK9 inhibitors. JACC Case Rep. 2025;30:103696.
130. Tardif JC, Pfeffer MA, Kouz S, et al. Pharmacogenetics-guided dalcetrapib therapy after an acute coronary syndrome: the dal-GenE trial. Eur Heart J. 2022;43:3947-56.
131. Puri R, Nissen SE, Arsenault BJ, et al. Effect of C-reactive protein on lipoprotein(a)-associated cardiovascular risk in optimally treated patients with high-risk vascular disease: a prespecified secondary analysis of the ACCELERATE trial. JAMA Cardiol. 2020;5:1136.
132. Centa M, Ketelhuth DF, Malin S, Gisterå A. Quantification of atherosclerosis in mice. J Vis Exp. 2019;148:e59828.
133. Barettino A, González-Gómez C, Gonzalo P, et al. Endothelial YAP/TAZ activation promotes atherosclerosis in a mouse model of Hutchinson-Gilford progeria syndrome. J Clin Investig. 2024;134:e173448.
134. Hamczyk MR, Nevado RM, Gonzalo P, et al. Endothelial-to-mesenchymal transition contributes to accelerated atherosclerosis in hutchinson-gilford progeria syndrome. Circulation. 2024;150:1612-30.
135. Damluji AA, Nanna MG, Mason P, et al. Coronary artery revascularization in the older adult population: a scientific statement from the American Heart Association. Circulation. 2025;152:25.
136. Jin KT, Du WL, Lan HR, et al. Development of humanized mouse with patient‐derived xenografts for cancer immunotherapy studies: a comprehensive review. Cancer Sci. 2021;112:2592-606.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright

Data & Comments
Data
0











Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].