


Precision medicine in metabolic dysfunction-associated steatotic liver disease: genetics, epigenetics, and emerging therapies
 0
0 Abstract
Metabolic dysfunction-associated steatotic liver disease (MASLD) is a major driver of chronic liver disease and an increasingly important etiology for hepatocellular carcinoma (HCC). Despite shared metabolic risk factors, MASLD shows marked inter-individual heterogeneity in fibrosis progression, treatment response, and HCC risk, indicating that one-size-fits-all management is insufficient. This narrative review synthesizes advances in precision medicine for MASLD with emphasis on (i) genetic risk stratification (major variants such as PNPLA3 and TM6SF2, protective alleles such as HSD17B13/MARC1, polygenic risk scores (PRS), and ancestry-specific effects); (ii) functional genomics and cell biology [bulk and single-cell expression quantitative trait locus (eQTL) mapping, spatial multi-omics, and cell-state/zonation biology]; and (iii) epigenetic regulation (DNA methylation, histone modifications, and microRNA networks) linking environment to phenotype. We discuss translational implications including biomarker-enabled trial enrichment, risk-based surveillance for advanced fibrosis and HCC, and emerging therapeutics ranging from liver-directed small interfering RNA/antisense oligonucleotides to epigenetic modulators. Finally, we outline how multimodal data integration and artificial intelligence may support clinically deployable risk models and treatment selection. Collectively, the field is moving from population-based management toward individualized prevention and therapy for MASLD and its complications.
Keywords
INTRODUCTION
Metabolic dysfunction-associated steatotic liver disease (MASLD), formerly termed nonalcoholic fatty liver disease (NAFLD), is now the most prevalent chronic liver disease worldwide and a growing contributor to cirrhosis, liver-related mortality, and hepatocellular carcinoma (HCC), and it is emerging as a leading indication for liver transplantation[1,2]. The recently adopted steatotic liver disease (SLD) nomenclature underscores that metabolic dysfunction is central to pathogenesis and clinical risk, moving the field away from exclusion-based definitions[3,4]. As the prevalence of obesity and type 2 diabetes mellitus increases globally, MASLD has become a major priority in hepatology, gastroenterology, and primary care, with an expanding need for risk stratification and precision prevention of advanced fibrosis and HCC[5].
MASLD’s clinical spectrum extends from isolated hepatic steatosis to metabolic dysfunction-associated steatohepatitis (MASH), progressive fibrosis, cirrhosis, and HCC, with advanced fibrosis associated with increased all-cause and liver-related mortality[6,7]. Beyond clinical implications, the economic burden is staggering: annual direct healthcare costs in the United States are projected to escalate from $103 billion to over $1 trillion within the next decade[8]. This economic strain is compounded by rising MASH-related transplantation rates and costly management of extrahepatic manifestations, including cardiovascular disease and malignancy[9-11]. These converging clinical and economic challenges underscore the importance of mechanistically informed risk stratification and personalized intervention strategies.
The molecular pathogenesis of MASLD follows a “multiple-parallel hits” model wherein insulin resistance, lipotoxicity, oxidative stress, dysbiosis, and genetic predisposition concurrently drive progressive hepatic injury[12-14]. Although numerous therapeutic agents targeting MASH pathways - including thyroid hormone receptor-β agonists (e.g., resmetirom) and incretin-based therapies (e.g., semaglutide, tirzepatide) - are actively being developed or have been recently approved[15-17], significant challenges, including long-term efficacy, tolerability, and adverse event profiles, particularly gastrointestinal side effects and sarcopenia, remain[18,19]. These obstacles largely reflect MASLD’s intricate pathogenetic heterogeneity, which varies substantially based on an individual’s genetic background, environmental exposure, and epigenetic state.
Furthermore, the recent adoption of the SLD nomenclature has highlighted the clinical heterogeneity among SLD subtypes - including MASLD, metabolic dysfunction and alcohol-related liver disease (MetALD), and alcohol-associated liver disease (ALD) - each of which exhibits distinct clinical trajectories and fibrotic progression patterns, thereby requiring subtype-specific management approaches[20]. A comprehensive understanding of the molecular intricacies of MASLD pathogenesis, coupled with precision-stratified approaches, is imperative for achieving superior therapeutic outcomes. In this study, we review cutting-edge discoveries in MASLD genetics, epigenetics, and multi-omics, emphasizing population-specific genetic variations and the translation of molecular insights into precision therapeutic strategies. We focus on Asian population studies, emerging gene-directed therapies, disease-specific and single-cell expression quantitative trait loci (eQTL) discoveries, and the integration of artificial intelligence (AI) in biomarker development and clinical trial design.
GENETIC ARCHITECTURE OF MASLD
Major risk variants and polygenic architecture
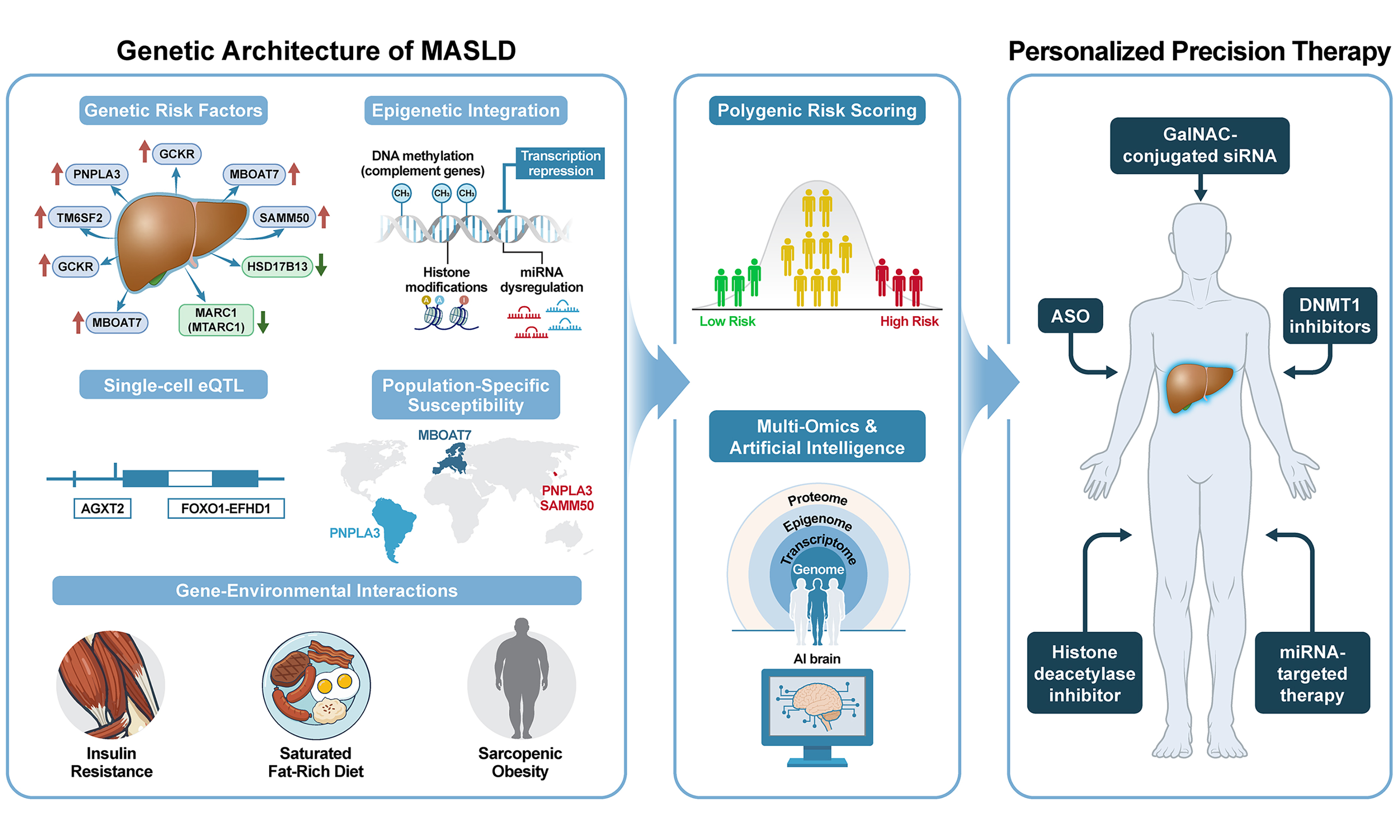
Understanding MASLD’s genetic architecture is essential for deciphering the disease’s clinical heterogeneity and individual risk of progression. Genome-wide association studies (GWAS) have consistently identified several variants showing robust associations with MASLD susceptibility and disease severity. Key loci include patatin-like phospholipase domain-containing 3 (PNPLA3), transmembrane 6 superfamily member 2 (TM6SF2), sorting and assembly machinery component 50 homolog (SAMM50), glucokinase regulator (GCKR), membrane-bound O-acyltransferase 7 (MBOAT7), and protective hydroxysteroid 17-beta dehydrogenase 13 (HSD17B13) variants. These genetic determinants can be broadly classified into hepatic fat-promoting risk alleles and protective variants, each modulating distinct lipid and inflammatory pathways[21,22]. Complementing traditional GWAS approaches, whole-exome sequencing coupled with machine learning algorithms has enabled the discovery of rare variants associated with severe phenotypes[21,22], thus establishing a comprehensive genomic foundation for precision risk stratification and targeted gene therapy development [Table 1].
Key genetic variants in MASLD
| Gene | Key variant (rsID) | Harmful or protective | Alteration in function | Disease contribution | Ethnic variations |
| PNPLA3 | rs738409 C>G (I148M) | Harmful | Loss of triglyceride lipase activity; promotes hepatocyte lipid retention[23] | MASLD, advanced fibrosis, HCC[24] | Highest frequency in Hispanics; intermediate in Europeans; lowest in individuals of African ancestry[25] |
| TM6SF2 | rs58542926 C>T (E167K) | Harmful | Reduces VLDL secretion from hepatocytes; increases hepatic triglyceride content[26] | Severe steatosis, fibrosis, cirrhosis[27,28] | More frequent in Europeans[29] |
| GCKR | rs1260326 C>T (P446L) | Harmful | Increases glucokinase activity; leads to hepatic glucose uptake and de novo lipogenesis[30] | Steatosis, hypertriglyceridemia[31] | |
| MBOAT7 | rs641738 C>T | Harmful | Alters phospholipid membrane composition and inflammation[32,33] | MASH, fibrosis[33-35] | Highest association in European-Caucasians[35] |
| SAMM50 | rs738491 | Harmful | Impairs mitochondrial outer membrane protein assembly; alters fatty acid oxidation[36] | MASLD, MASH, fibrosis[37] | Strongest association in East Asians (including Korean)[38-40] |
| HSD17B13 | rs72613567:TA | Protective | Loss of function; promotes fat breakdown[41,42] | Lower necroinflammation, MASH, fibrosis, HCC risk[43,44] | |
| MARC1 | rs2642438, p.A165T | Protective | Reduce intracellular neutral lipid content[45] | Lower liver fat (MASLD) and inflammation[46] |
PNPLA3 I148M: the dominant risk allele
The PNPLA3 rs738409 C>G (I148M) variant is the single most influential risk allele for MASLD, showing robust associations with hepatic fat content, lobular inflammation, hepatocellular ballooning, advanced fibrosis, and HCC across diverse populations[24]. Mechanistically, the I148M substitution impairs triglyceride hydrolase activity, causing mutant PNPLA3 protein accumulation on lipid droplet surfaces and obstruction of normal lipolysis, thereby amplifying lipotoxic hepatocellular stress[23]. Although these associations are robustly replicated, the reported effect estimates, particularly for HCC risk, vary substantially across cohorts, likely reflecting differences in alcohol co-exposure, ancestry, and underlying liver disease etiology[25].
TM6SF2 E167K and hepatic lipoprotein metabolism
TM6SF2 rs58542926 C>T (E167K) is the second major locus, with carriers exhibiting elevated hepatic triglyceride content but reduced circulating low-density lipoprotein (LDL) and very-low-density lipoprotein (VLDL) cholesterol. These phenotypes suggest compromised efficiency of hepatic lipoprotein secretion[26]. Clinically, this variant is associated with more severe steatosis and an elevated risk of advanced fibrosis and cirrhosis[27,28]. When combined with PNPLA3 risk variants, TM6SF2 identifies a high-risk MASLD subgroup with a pronounced hepatic disease burden[47,48].
GCKR, MBOAT7, and metabolic modulators
Variants in GCKR and MBOAT7 modulate MASLD susceptibility by altering carbohydrate and phospholipid metabolism, respectively. GCKR rs1260326C>T (P446L) and related functional variants enhance glucokinase activity, thereby increasing hepatic glucose uptake and de novo lipogenesis (DNL), predisposing to steatosis and hypertriglyceridemia, particularly in insulin-resistant individuals[30,31]. MBOAT7 rs641738 C>T reduces lysophosphatidylinositol acyltransferase expression, disrupting membrane phospholipid composition, promoting inflammatory signaling, and increasing vulnerability to steatohepatitis and fibrosis progression[33,34]. SAMM50 rs738491 - encoding an outer mitochondrial membrane protein essential for mitochondrial morphology - modulates reactive oxygen species (ROS) levels and β-oxidation capacity[36,37] and demonstrates a significant association with MASLD in the Korean GWAS[38].
HSD17B13: a protective allele
In contrast to risk alleles, HSD17B13 rs72613567:TA confers robust protection against progression from isolated steatosis to steatohepatitis, advanced fibrosis, and HCC[41,43]. Carriers demonstrate lower necroinflammatory activity and milder fibrosis via attenuation of lipotoxic injury[44]; mechanistically, HSD17B13 phosphorylation at S33 promotes hepatic fat catabolism, whereas the S33A mutation impairs this protective function, thereby promoting steatosis and MASH severity[42].
MARC1 (MTARC1): a protective variant
Beyond HSD17B13, MARC1 (also known as MTARC1) has emerged as another genetically validated protective locus in MASLD. A common missense variant (rs2642438, p.A165T) in MARC1, first identified through GWAS for all-cause liver cirrhosis, confers protection against hepatic steatosis, cirrhosis, and HCC[46]. The protective p.T165 allele results in reduced MARC1 protein stability and increased proteasomal degradation, leading to approximately 50% lower hepatic MARC1 protein levels[45].
PRS for precision stratification
Individual risk variants can be aggregated into PRS, capturing cumulative genetic liability for advanced MASLD phenotypes[49]. Simple PRS incorporating PNPLA3, TM6SF2, and HSD17B13 significantly improve prediction of significant fibrosis and histologic severity beyond clinical factors alone in population-based cohorts[50,51]. More sophisticated partitioned PRS approaches, which dissect “liver-centric” vs. “cardiometabolic” genetic components, have recently identified distinct MASLD subtypes with divergent patterns of hepatic fibrosis, HCC risk trajectories, and systemic cardiometabolic complications, providing a molecular framework for precision endotyping[52]. However, most current PRS validation studies are cross-sectional, and prospective evidence demonstrating the clinical utility of risk-based interventions is still emerging.
Disease-specific and single-cell eQTL mapping: beyond GWAS
While GWAS identify risk loci, eQTL mapping provides the mechanistic link between genetic variants and their functional consequences in gene expression. Conventional eQTL approaches using healthy liver tissues have profiled thousands of gene-regulatory variants; however, they fail to capture dynamic regulatory effects relevant to specific disease contexts[53,54].
Yoo et al. devised a disease-specific eQTL screening pipeline using liver biopsies and blood samples from 125 Korean individuals (83 with biopsy-proven MASLD and 42 without MASLD)[53]. The authors identified 242 MASLD-specific eQTLs, which are genetic variants whose regulatory effects on gene expression are detectable only in the diseased liver environment, not under healthy conditions, through eQTL analyses of 21,272 transcripts and over 3.2 million genotyped/imputed SNPs. Among these, alanine-glyoxylate aminotransferase 2 (AGXT2) emerged as a top MASLD-eQTL: the eSNP rs2291702 constituted a significant cis-eQTL specifically in the MASLD group (P = 7.21 × 10-9) but not in the no-MASLD controls (P = 0.38) or GTEx liver data (P = 0.31). Notably, AGXT2 downregulation was restricted to rs2291702:CC homozygotes within the MASLD group, demonstrating genotype-dependent gene regulation under disease conditions. This finding was replicated in an independent cohort of 162 Korean individuals (combined P = 8.05 × 10-8). Functional validation confirmed that AGXT2 knockdown exacerbated MASLD diet-induced liver fibrosis by increasing endoplasmic reticulum (ER) stress-mediated hepatocellular death, whereas AGXT2 overexpression attenuated fibrosis and steatosis[53]. These results established AGXT2 as a genotype-dependent therapeutic candidate and demonstrated that disease-specific eQTL mapping could uncover regulatory variants not detected by conventional approaches.
Building on this framework, Hong et al. advanced the approach to single-cell resolution by performing single-nucleus RNA sequencing (snRNA-seq) on liver biopsies from 25 patients with MASLD and 23 controls[54]. This single-nucleus eQTL analysis, employing a Poisson mixed-effects model, identified over 3,500 liver-eQTLs across four major liver cell types [hepatocytes, cholangiocytes, hepatic stellate cells (HSCs), and endothelial cells], with the majority being cell type-specific (58.3%-72.8%). A critical innovation was the identification of cell-state-interacting eQTLs (ieQTLs), which are genetic regulatory effects whose magnitude varies depending on the cell’s functional state or zonal position within the hepatic lobule. Strikingly, ieQTLs demonstrated significant enrichment for MASLD trait heritability [enrichment odds ratio (OR) = 10.27], substantially exceeding that of conventional liver-eQTLs (OR = 5.39). Among the most notable ieQTLs, the protective variant rs28636836 in HSD17B13 exhibited zone-specific regulation with the strongest eQTL effect in periportal hepatocytes, while CYP4V2, a druggable fatty acid metabolism gene, showed periportal-specific regulation. These zonation-dependent eQTLs have immediate implications for patient stratification in RNA-based therapeutic trials and for the discovery of zone- and genotype-specific treatment targets.
The study further integrated transcription factor (TF) activity analysis with iQTLs, revealing 601 functional “quartets” comprising TFs, cell states, ieSNPs, and ieGenes. Based on this framework, the investigators pinpointed the FOXO1-rs13395911-EFHD1 axis: in maladapted hepatocytes (characterized by reduced Hep-M12 metabolic module expression), decreased FOXO1 activity led to the loss of EFHD1 expression specifically in rs13395911:TT genotype carriers[54]. EFHD1 protected against lipotoxicity-induced ER stress, mitochondrial dysfunction, and apoptosis in cell lines and human hepatic organoids, suggesting that it is a genotype-dependent therapeutic target for MASLD.
POPULATION-SPECIFIC GENETIC SUSCEPTIBILITY
Ethnic heterogeneity in variant distribution
Genetic risk factors for MASLD exhibit profound ethnic heterogeneity, necessitating localized genomic research to develop genomically and ethnicity-adapted clinical management strategies. The global distribution of key risk alleles varies markedly, directly influencing disease prevalence and severity across populations.
The PNPLA3 rs738409 C>G variant exemplifies striking ethnic disparities, with the risk G allele reaching approximately 50% frequency in Hispanics of Mexican, Central, or South American descent, correlating with their disproportionately high rates of MASH, advanced fibrosis, and HCC[25]. Conversely, African and African-American populations exhibit the lowest G-allele frequency (AF) (approximately 12%-14%), conferring protection against hepatic steatosis despite elevated systemic insulin resistance and obesity burdens. Individuals of European ancestry occupy an intermediate frequency range (approximately 23%), showing moderate MASLD susceptibility.
Population-specific variant patterns
TM6SF2 rs58542926 C>T contributes to ethnic variation: prevalence reaches 7.2%, 4.7%, and 3.4% in individuals of European ancestry, Hispanics, and African-Americans, respectively, with variant carriers exhibiting increased hepatic triglyceride retention due to impaired VLDL secretion[29]. MBOAT7 rs641738 C>T shows the strongest associations with MASLD progression and fibrosis in European-Caucasian cohorts, while demonstrating inconsistent-to-absent associations in Asian and African-American populations[35,55].
Lean MASLD and genetic dominance
Meta-analysis has revealed that lean MASLD prevalence [defined as body mass index (BMI) < 25 kg/ m2 in Caucasians or BMI < 23 kg/ m2 in Asians] was 12% in Asia, 10.2% in the Middle East, and 9.2% in Western countries[56]. In lean MASLD cohorts, genetic predisposition - particularly PNPLA3 and SAMM50 variants - predominates over systemic adiposity in driving MASLD, explaining MASLD occurrence in lean individuals independent of manifest metabolic syndrome[39]. This ancestry-driven genetic architecture necessitates ethnicity-specific predictive models extending beyond traditional obesity paradigms.
Korean population-specific insights
Within Korean populations, the GENIE cohort study established significant associations between PNPLA3 and SAMM50 genes and both MASLD presence and severity[40]. Additional GWAS identified associations of PNPLA3 with metabolic dysfunction-associated fatty liver disease, specifically with overweight/obese phenotypes[57]. PNPLA3 variants are correlated with increased immune cell infiltration and advanced fibrosis progression[58]. Notably, a disease-specific eQTL approach applied to the Korean biopsy-proven MASLD cohort revealed population-specific regulatory dynamics: the AGXT2 eSNP rs2291702 showed variable AFs across populations, with the reference T allele predominating in African populations (mean AF = 0.802) but being minor in East Asians and Koreans (AF = 0.327 and 0.330, respectively), implying differential susceptibility to the AGXT2-dependent MASLD pathway across ethnicities[53]. These Korean-specific genetic discoveries collectively support the development of ancestry-tailored PRS and precision screening strategies targeting the evolving MASLD burden in East Asian populations.
Global implications and cross-ancestry considerations
Although the Korean cohort findings summarized above provide robust East Asian-specific evidence, they should be interpreted within the broader global landscape of MASLD genetic susceptibility. The disproportionate burden of MASH, advanced fibrosis, and HCC in Hispanic populations is largely attributable to the high frequency of the PNPLA3 I148M risk allele (approximately 50%)[25], whereas individuals of African ancestry exhibit relative protection from hepatic steatosis despite the high prevalence of obesity and insulin resistance, partly owing to the low frequency of the PNPLA3 risk allele[25,29]. South Asian, Middle Eastern, and African populations remain markedly underrepresented in MASLD genomic studies despite the rapidly rising disease burden[5]. Consequently, ancestry-diverse genomic studies and population-specific validation of risk variants and PRS are essential to ensure that emerging precision medicine approaches are equally applicable across global populations, rather than disproportionately benefiting individuals of European ancestry.
EPIGENETIC MODIFICATIONS IN MASLD
DNA methylation landscape
Epigenetic regulation - encompassing DNA methylation, histone modifications, and non-coding RNAs - bridges environmental exposures (diet, obesity, insulin resistance, dysbiosis) with heritable alterations in gene expression driving MASLD initiation, progression, and potential reversal. DNA methylation patterns dynamically track disease progression from steatosis through fibrosis, with epigenome-wide association studies (EWAS) identifying differentially methylated regions that are strongly correlated with fatty liver index and capture longitudinal changes in steatosis burden[59].
Korean liver biopsy EWAS (n = 106) identified hypermethylation and downregulation of complement genes (C1R, C1S, C3, C6, C4BPA, SERPING1) alongside hypomethylation and upregulation of C5AR1, C7, and CD59, patterns that correlate with MASLD histological severity (steatosis, inflammation, fibrosis). These findings were validated in public datasets (GSE180474) and MASH mouse models[60]. Recent whole-genome methylation sequencing in MASLD cohorts identified sex-specific DNA methylation signatures and circulating cell-free DNA biomarkers (e.g., TM4SF5), facilitating highly accurate non-invasive fibrosis stage prediction[61]. These observations underscore DNA methylation as a pivotal epigenetic marker that bridges genetic susceptibility with environmental factors, providing a framework for precision medicine through non-invasive diagnostics and personalized longitudinal monitoring across the MASLD spectrum.
Beyond descriptive methylome profiling, recent mechanistic studies have directly implicated DNA methyltransferase 1 (DNMT1) as a central epigenetic driver of MASLD progression. Sohn et al. conducted integrated analyses of the hepatic transcriptome (n = 131) and DNA methylome (n = 106) from patients with biopsy-confirmed MASLD and demonstrated consistent upregulation of DNMT1 expression across multiple independent MASLD cohorts, correlated positively with histological severity, including hepatocellular ballooning, lobular inflammation, NAFLD activity score (NAS), and fibrosis stage[62]. DNMT1 expression was positively correlated with insulin resistance parameters (homeostatic model assessment for insulin resistance, circulating insulin, HbA1c), linking epigenetic dysregulation to metabolic perturbation. A meta-analysis integrating multiple cohorts confirmed reproducible DNMT1 upregulation, whereas DNMT3A and DNMT3B did not show consistent changes[62]. Although the reproducibility of DNMT1 upregulation across multiple cohorts is relatively well-established, several other epigenetic observations summarized in this section, including specific differentially methylated regions and chromatin accessibility signatures, are based on single-cohort data and require independent validation.
DNA methylome profiling in both human MASLD samples and a diet-induced mouse model revealed significant enrichment of binding motifs of key hepatic TFs, particularly hepatocyte nuclear factor 4α (HNF4α) and peroxisome proliferator-activated receptor α (PPARα), among MASLD-associated hypermethylated differentially methylated positions[62]. Importantly, the expression of HNF4α and PPARα was inversely correlated with histological indicators of disease activity (ballooning, lobular inflammation) and with DNMT1 expression in human MASLD samples, indicating that DNMT1-mediated hypermethylation functionally suppressed these master metabolic regulators during disease progression.
Histone modifications and chromatin remodeling
Histone modifications, such as acetylation and methylation, are important epigenetic processes controlling gene expression in chronic diseases, such as MASLD fibrosis. These reversible histone marks dynamically regulate profibrogenic gene expression in HSCs, the primary mediators of extracellular matrix deposition and fibrosis progression[63].
Complementing studies on histone modification, genome-wide chromatin accessibility profiling using the assay for transposase-accessible chromatin with sequencing (ATAC-seq) has provided a comprehensive epigenomic reference for MASLD progression. Kang et al. performed ATAC-seq on 44 liver biopsy samples spanning the full histological spectrum of MASLD - from healthy controls through isolated steatosis to fibrotic MASH - in a Korean biopsy-proven cohort[64]. A total of 112,303 merged accessible chromatin regions were identified. Principal component analysis and correlation heatmaps clearly distinguished disease stages by their chromatin accessibility profiles.
Compared with isolated steatosis, fibrotic MASH showed 272 up-regulated and 1,137 down-regulated differentially accessible regions (DARs) (false discovery rate < 0.05)[64]. Functional enrichment analysis of down-regulated DARs in fibrotic MASH revealed significant associations with hormone response, cell cycle regulation, steroid metabolic processes, and liver development pathways. TF binding motif enrichment and de novo motif analyses within these DARs identified candidate biomarkers, including ETS family members (ETV1, ETV4, ELF3, GABPA), EWSR1-FLI1, IKZF1, SPI1, STAT2, and ZNF384, which could serve as epigenomic diagnostic markers distinguishing MASLD progression stages[64]. These chromatin accessibility signatures complement DNA methylation and histone modification data, collectively delineating the regulatory specificity of non-coding genomic regions affected by genetic and epigenetic alterations during MASLD progression.
MicroRNA networks and post-transcriptional regulation
MicroRNAs (miRNAs) orchestrate MASLD pathogenesis through post-transcriptional modulation of lipid homeostasis, inflammatory signaling, and extracellular matrix remodeling. Liver-enriched miR-122, comprising approximately 70% of total hepatic miRNAs, regulates cholesterol biosynthesis and fatty acid metabolism. Circulating miR-122 levels increase in steatosis and MASH due to hepatocyte damage, functioning as a sensitive biomarker[65,66]. miR-192, extensively studied alongside miR-122, is upregulated in MASH relative to isolated steatosis, contributing to disease progression through inflammation modulation (e.g., M1 macrophage polarization via Rictor/Akt/FoxO1 signaling)[67]. Circulating miRNA panels provide non-invasive diagnostic, staging, and monitoring capabilities for MASLD/MASH, surpassing single-marker performance through multi-miRNA combinations[68,69]. Therapeutic targeting of miRNAs, particularly miR-122 via antisense inhibition, reduces hepatic lipid accumulation, inflammation, and fibrosis in MASLD models, with demonstrated clinical translation in hepatitis C virus trials and emerging MASLD applications[70].
GENE-ENVIRONMENT INTERACTIONS
Systemic metabolism, diet, and obesity in MASLD pathogenesis
Gene-environment interactions are key to understanding heterogeneous MASLD phenotypes and variable progression rates among individuals with similar metabolic risk factors. Disease expression reflects a dynamic interplay between inherited susceptibility alleles and modifiable environmental exposures, including systemic metabolic status, dietary patterns, adiposity distribution, and physical activity levels[71,72].
Hepatic and peripheral insulin resistance represent major upstream drivers of MASLD. Insulin resistance amplifies adipose tissue lipolysis, delivering excess free fatty acids to the liver and stimulating DNL, thereby promoting intrahepatic triglyceride accumulation and lipotoxic injury[73]. High-carbohydrate diets rich in fructose and refined sugars enhance DNL by upregulating ChREBP and SREBP1c TFs, while simultaneously promoting oxidative stress and gut-derived endotoxemia that aggravate steatohepatitis[74,75]. Saturated fat-rich diets increase hepatocellular ceramides and diacylglycerols, impairing insulin signaling and amplifying inflammatory and fibrogenic responses[76,77]. Obesity is a potent pro-inflammatory amplifier owing to adipokine secretion (elevated leptin and resistin; reduced adiponectin), which creates an inflammatory milieu that accelerates the transition from isolated steatosis to MASH and fibrosis[78]. Sarcopenic obesity, whose prevalence in aging populations and Asian cohorts is increasing, compounds the risk of MASLD/MASH progression through reduced skeletal muscle glucose disposal capacity and altered myokine secretion. This worsens systemic insulin resistance and inflammation[79,80]. Physical inactivity aggravates these pathways, whereas higher moderate-to-vigorous physical activity levels are associated with lower steatosis and fibrosis risk, independent of genetic and metabolic risk factors[81,82].
Genotype-dependent modulation of environmental effects
The magnitude of MASLD induced by metabolic and dietary stressors is substantially modified by individual genotype. PNPLA3 rs738409 G (I148M) allele carriers exhibit more severe hepatic steatosis than non-carriers for equivalent adiposity levels, with genetic effects amplified by sugar-sweetened beverage consumption[83]. PNPLA3 G-allele carriers experiencing ≥ 10 kg weight gain after early adulthood demonstrate markedly elevated MASLD prevalence even at non-obese BMI, illustrating the combined impact of genetic susceptibility and modest weight gain on disease expression[84].
Similarly, TM6SF2 rs58542926 T-allele carriers exhibit reduced hepatic VLDL-triglyceride export, causing caloric and fat excess to preferentially manifest as intrahepatic triglyceride and cholesterol retention with amplified fibrosis risk despite relatively favorable circulating lipid profiles[28,85]. MBOAT7 risk alleles alter hepatocellular phosphatidylinositol acyl-chain composition, enhancing pro-inflammatory signaling and HSC activation, thereby increasing fibrosis susceptibility for equivalent obesity and insulin resistance burdens[86,87].
Genotype-specific responses to lifestyle interventions
Gene-environment interactions extend to variable treatment responses to lifestyle modification. While weight reduction, Mediterranean-style dietary patterns, and lower-carbohydrate regimens remain foundational strategies for most patients, genetic profiling helps explain inter-individual variation in intervention efficacy[72]. Depending on inherited polygenic risk, some individuals benefit from modest dietary and physical activity improvements, whereas others who particularly do not respond to metabolic targeted therapies [e.g., thyroid hormone receptor beta (THR-β) agonists and glucagon-like peptide-1 (GLP-1) receptor agonists] require more intensive integrated programs combining tailored nutrition, structured exercise, and emerging gene- and epigenome-targeted therapies for truly personalized MASLD management.
FROM MOLECULAR MECHANISMS TO THERAPEUTIC TARGETS
Genetic and epigenetic drivers of MASLD are increasingly being elucidated, and therapeutic strategies are evolving beyond nonspecific metabolic control (e.g., weight loss, insulin sensitizers, THR-β agonists, and GLP-1 receptor agonists) toward interventions that directly modulate causal molecules and pathways. Gene-based and epigenome-targeted approaches represent emerging therapeutic focuses [Figure 1]. Importantly, the early-phase clinical trials reviewed below represent the first concrete clinical implementation of precision medicine in MASLD, as they enroll participants based on genotype rather than histological phenotype alone. Hepatocyte-targeted siRNA and ASO programs against PNPLA3 I148M (e.g., JNJ-75220795, AZD2693, ALN-PNP, and LY3849891) recruit homozygous risk-allele carriers, while RNA interference trials targeting HSD17B13 (e.g., ARO-HSD and ALN-HSD) phenocopy a genetically protective phenotype[88-91]. Although most of these programs are still in phase I/II and clinically meaningful improvements in fibrosis endpoints have not yet been consistently demonstrated, they collectively establish the operational feasibility of genotype-enriched MASLD trials and provide a template for future genotype-guided therapeutic development.
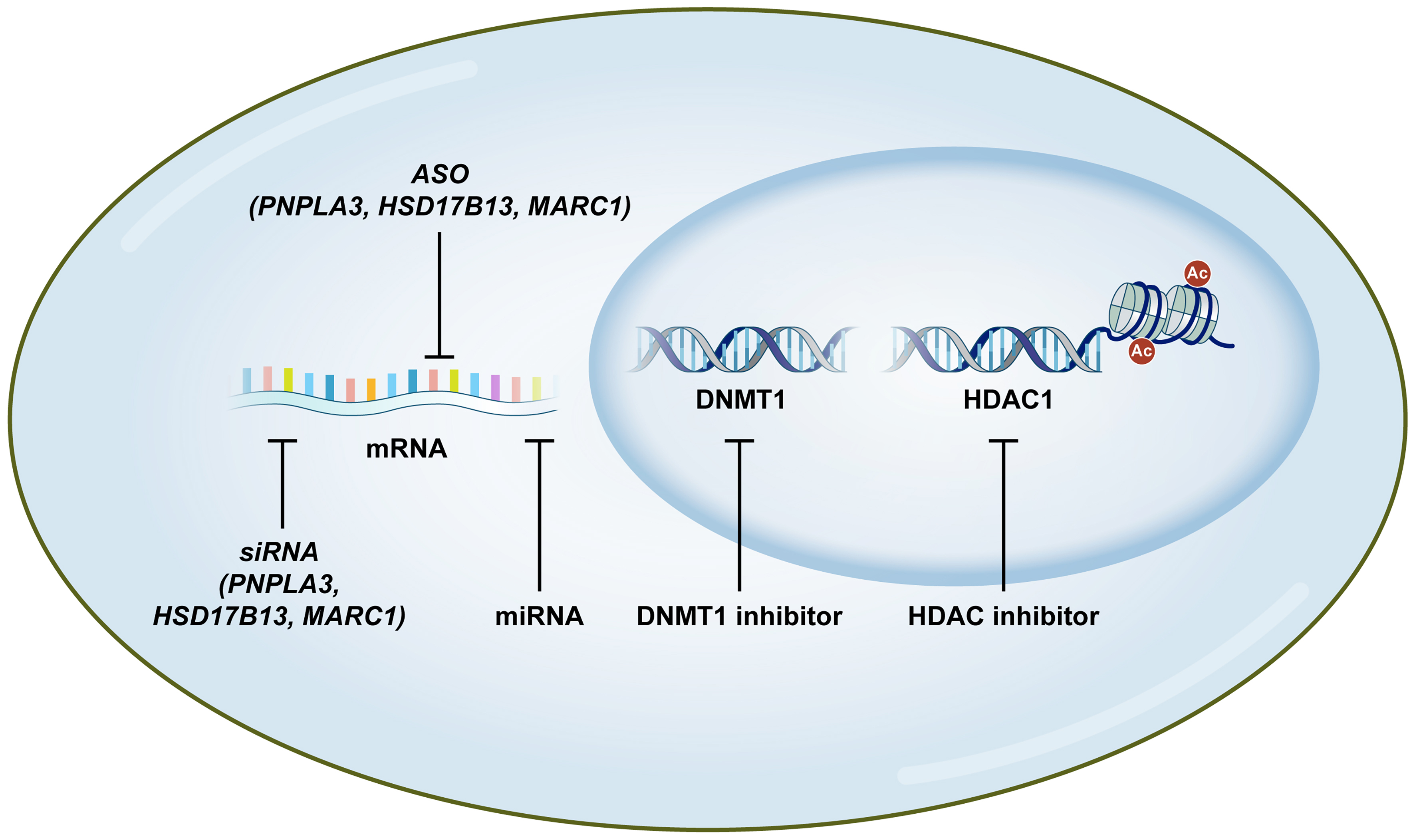
Figure 1. Emerging genetic and epigenetic therapeutic strategies for MASLD. The schematic illustrates the multi-layered precision medicine approaches targeting MASLD at various molecular levels. Nucleotide-based therapeutics, including siRNA and ASO, specifically target and silence mRNA of major genetic variants such as PNPLA3, HSD17B13, and MARC1 to reduce toxic gain-of-function or modulate disease progression. Additionally, miRNA-targeted agents can reset pathogenic transcriptional networks. Within the nucleus, epigenetic modulators such as DNMT1 inhibitors and HDAC inhibitors reprogram aberrant transcriptional states by reversing DNA hypermethylation and histone deacetylation, thereby restoring the expression of master metabolic regulators. Ac: acetylation; ASO: antisense oligonucleotide; DNMT1: DNA methyltransferase 1; HDAC1: histone deacetylase 1; HSD17B13: hydroxysteroid 17-beta dehydrogenase 13; MARC1: mitochondrial amidoxime reducing component 1; MASLD: metabolic dysfunction-associated steatotic liver disease; miRNA: microRNA; mRNA: messenger RNA; PNPLA3: patatin-like phospholipase domain-containing 3; siRNA: small interfering RNA.
Genetic therapeutic approaches
Gene-based therapy encompasses a diverse spectrum of strategies tailored to specific therapeutic objectives, ranging from transient gene modulation to permanent genomic correction. Transient approaches such as small-interference RNA (siRNA) and antisense oligonucleotides (ASOs) provide reversible control over malfunctioning genes[92]. These evolving technologies - including high-precision base and prime editing - are currently being leveraged to restore metabolic homeostasis in liver diseases, with several clinical trials evaluating the suppression of key genetic targets to treat MASLD underway.
siRNA targeting PNPLA3 and hepatic genes
PNPLA3 I148M represents an archetypal “druggable” genetic driver, because the mutant protein accumulates on lipid droplets, mediating gain-of-toxic-function effects without systemic benefit. In preclinical PNPLA3 I148M-driven MASH models, hepatocyte-targeted, minor-allele-specific N-acetylgalactosamine (GalNAc)-conjugated siRNA against PNPLA3 achieved robust mutant protein knockdown, reduced hepatic triglyceride content, inflammatory and profibrotic gene expression, and improved histologic steatosis and inflammation without overt hepatic or extrahepatic toxicity signals[93].
GalNAc-conjugated hepatocyte-targeted PNPLA3 siRNA (JNJ-75220795)underwent phase I evaluation in PNPLA3 I148M homozygotes, where single subcutaneous dosing produced dose-dependent liver fat reductions up to 46% (measured by magnetic resonance imaging) sustained for ≥ 24 weeks, with no major adverse events documented during follow-up[88]. ALN-PNP and LY3849891 are additional early-phase GalNAc-siRNA candidates targeting PNPLA3-driven pathogenic pathways, aimed at reducing hepatic fat and improving metabolic parameters[94,95]. GalNAc-conjugated siRNA against HSD17B13 is promising: dosing achieved dose-dependent hepatic HSD17B13 mRNA knockdown with favorable trends in levels of liver enzymes and histologic activity in phase I MASH trials, with acceptable safety profiles[89]. Hepatocyte-targeted siRNA ARO-HSD in additional phase I/II MASH studies demonstrated robust HSD17B13 mRNA and protein reduction with accompanying improvements in alanine aminotransferase and good short-term tolerability[90]. Despite robust target engagement and favorable short-term safety, clinically meaningful improvements in fibrosis endpoints have not yet been consistently demonstrated across these early phase trials, and long-term efficacy and safety data are awaited.
ASO strategies
Preclinical studies on GalNAc-conjugated ASO demonstrated that silencing the PNPLA3 I148M variant effectively ameliorated liver inflammation and fibrosis[96]. Phase I evaluation of AZD2693 demonstrated favorable safety and robust target engagement - evidenced by 89% reduction in hepatic mRNAs - while achieving dose-dependent liver fat decreases and improvements in circulating inflammatory markers in homozygous risk-allele carriers[91]. Notably, ASO targeting HSD17B13 in CDAHFD-induced MASH models significantly modulated hepatic steatosis but failed to yield substantial fibrosis improvement despite near-complete gene suppression. This highlights the complexity of monogenic targeting in polygenic disease.
MARC1 (MTARC1) as a therapeutic target
The genetically validated protective effect of MARC1 loss-of-function provides a strong rationale for its therapeutic targeting. Ciociola et al. demonstrated that MARC1 downregulation in primary human hepatocytes reduced neutral lipid content by enhancing β-oxidation, upregulated ferroptosis suppressor proteins, and reduced ROS levels, collectively suggesting a favorable therapeutic window[97]. The allele-specific effect - observed only in risk allele (p.A165) carriers and not in protective allele (p.T165) carriers - supports a precision medicine approach targeting MARC1 specifically in individuals carrying the risk allele. Beyond steatosis, Kovooru et al. showed that MTARC1 ablation in HCC cell lines suppressed key oncogenic pathways [epithelial-mesenchymal transition (EMT), hypoxia, epidermal growth factor receptor (EGFR), TGF-β signaling] and activated tumor suppressors (CDKN2A, fatty acid degradation via CPT1A), with in vivo xenograft tumors exhibiting markedly reduced growth (45% lower volume), vascularization, and proliferation (42% Ki-67 reduction)[98]. Importantly, the anti-tumor effects were causally linked to enhanced β-oxidation, as transient CPT1A knockdown partially reversed the benefits of MTARC1 knockout. These converging data from metabolic and oncologic studies position MARC1/MTARC1 as a therapeutic target for both MASLD prevention and MASLD-associated HCC, warranting investigation into the development of hepatocyte-targeted MARC1 inhibition strategies, such as GalNAc-conjugated siRNA or ASO approaches.
Epigenetic modulators
Epigenetic therapies aim to reprogram pathologic transcriptional states by targeting core mechanisms, including DNA methylation, histone modifications, and non-coding RNA-mediated gene regulation. In MASLD, accumulating evidence indicates that hepatocytes and HSCs acquire characteristic epigenetic alterations, including hypermethylation of lipid and cholesterol metabolism, more permissive chromatin at inflammatory and fibrogenic loci, and profoundly dysregulated miRNA networks[99]. Given the inherent reversibility of epigenetic marks, epigenetic drugs provide opportunities to “reprogram” hepatic transcriptional states and function without altering underlying DNA sequences.
DNA methyltransferase inhibitors
DNMT inhibitors, whose use has been established for hematologic malignancies, demethylate CpG islands to reactivate silenced genes linked to lipid export, insulin signaling, and antioxidant pathways, offering therapeutic potential for MASLD[99]. EWAS for progressive MASLD liver biopsies revealed gradual hypermethylation of metabolic genes and pathways (lipid metabolism, amino acid metabolism, PPAR signaling, complement/inflammation), which correlated with epigenetic age acceleration and histological severity (steatosis, hepatocellular ballooning, fibrosis), supporting DNMT-targeted reprogramming as a therapeutic avenue[100]. In Western diet-induced MASLD models, DNMT1 inhibitors (azacitidine and zebularine) restored macrophage autophagy by demethylating the LC3B, ATG5, and ATG7 promoters, thus shifting Kupffer cells from pro-inflammatory M1 to anti-inflammatory M2 phenotypes. Over 8 weeks, treatment improved insulin sensitivity and glucose tolerance, reduced steatosis, inflammation, and fibrosis, and normalized liver enzymes and lipid profiles without body weight changes[101].
More recently, Sohn et al. provided compelling evidence for DNMT1 as a selective therapeutic target using 5-aza-4’-thio-2’-deoxycytidine (Aza-TdC), a next-generation DNMT1-selective nucleoside analog[62]. In a diet-induced mouse model of MASLD, oral Aza-TdC administration during the final 4 weeks of an 18-week MASLD diet selectively depleted DNMT1 protein and attenuated MASLD progression, thus reducing hepatomegaly, hepatic lipid accumulation, macrophage infiltration, and NAS and fibrosis scores. Comprehensive lipidomic profiling demonstrated selective reduction of hepatic triacylglycerol species - predominantly those containing unsaturated fatty acid chains - while RNA-seq and snRNA-seq confirmed the downregulation of lipid uptake (Cd36), transport (Fabp1, Fabp4, Fabp7), and lipid droplet dynamics (Plin2, Plin3, Cidec) genes, as well as inflammatory markers (Adgre1, Tnfα, Il6, Tgfb1)[62]. Importantly, Aza-TdC reversed MASLD-associated hypermethylation at HNF4α- and PPARα-binding motif-containing regulatory regions, restoring the transcription of 42 target genes, including Pck1, Slc38a3, Il6ra, and Prlr. Chromatin immunoprecipitation sequencing validated the recovery of HNF4α occupancy and H3K27ac active histone marks at the Pck1 promoter, and CRISPRoff/on-based epigenome editing confirmed that targeted PCK1 promoter methylation directly modulated lipid accumulation in hepatocytes: CRISPRoff-mediated PCK1 silencing promoted triacylglycerol accumulation and upregulated lipogenic genes (SCD1, SREBP1, FASN, PLIN2), while CRISPRon-mediated demethylation restored PCK1 expression[62]. These data position selective DNMT1 inhibition as a mechanistically defined epigenetic therapeutic strategy for MASLD, targeting the restoration of HNF4α- and PPARα-dependent metabolic gene programs.
Histone deacetylase inhibitors
Histone deacetylase (HDAC) inhibitors represent a promising epigenetic therapy class that can reprogram aberrant hepatic transcriptional states by leveraging epigenetic mark reversibility. HDAC11-selective inhibition via compound B6 alleviates hepatic steatosis through LKB1/AMPK axis activation, simultaneously reducing DNL and enhancing fatty acid oxidation[102]. Beyond lipid metabolism, Class II HDAC inhibition effectively impairs HSC activation and liver fibrosis by promoting the anti-fibrotic miRNA-29[103]. Liver-targeting Class I selective inhibitors ensure high hepatic drug accumulation, offering potent and safer therapeutic strategies for advanced complications, including HCC[102]. These tissue-selective approaches provide a versatile framework for restoring metabolic and structural homeostasis across the MASLD spectrum.
MicroRNA-targeted therapies
miRNAs constitute a finely tuned regulatory layer integrating metabolic, inflammatory, and fibrogenic signals, making them conceptually appealing and mechanistically precise therapeutic targets in MASLD[70]. miRNA-directed agents can reset entire pathogenic networks underlying steatosis, steatohepatitis, and carcinogenesis through the modulation of a set of target transcripts rather than single proteins.
miR-122, the dominant liver-enriched miRNA and a key coordinator of cholesterol biosynthesis, fatty acid oxidation, and VLDL secretion, is downregulated in advanced MASLD and MASH; conversely, circulating levels increase and are biomarkers of hepatocyte injury[66]. Preclinical studies support hepatic miR-122 restoration via mimics to normalize lipid handling, dampen proliferation, and reduce HCC aggressiveness in steatotic and MASH-related carcinogenesis models[104].
Beyond miR-122, additional miRNAs are critical nodes in the MASLD pathogenic network owing to their coordination of multi-target metabolic responses. miR-10b-5p is reduced in diabetic and obese states, with systemic miR-10b-5p mimic delivery restoring KIT-KLF11 signaling, normalizing glucose homeostasis, and improving gastrointestinal motility across multiple diabetic mouse models, suggesting replacement therapy as a disease-modifying strategy that outperforms standard antidiabetic and prokinetic medications[105]. miR-132, consistently upregulated in MASLD/MASH, drives severe steatosis and hyperlipidemia in transgenic mice through coordinated metabolic target repression (FoxO3, PTEN, SIRT1); antisense miR-132 inhibition reverses hepatic steatosis and lowers LDL/VLDL levels, positioning anti-miR-132 as a promising network-level MASLD therapy[106]. Upregulation of miR-103/107 in obesity and fatty liver, with systemic silencing, improves glucose homeostasis, enhances insulin-stimulated glucose uptake in adipose tissue through caveolin-1 derepression and insulin receptor stabilization, and reduces adipocyte size, suggesting anti-miR-103/107’s potential for restoring insulin sensitivity in MASLD-associated metabolic dysfunction[107].
CLINICAL TRANSLATION AND PRECISION MEDICINE
Genetic risk assessment and stratification
PRS represents key tools that operationalize genomic discoveries for MASLD risk stratification and individualized clinical care. PRS aggregates common variant effects in genes regulating hepatic lipid levels (PNPLA3, TM6SF2, MBOAT7, GCKR, HSD17B13, MARC1) into a quantitative measure capturing inherited disease predisposition. De Vincentis et al. demonstrated that hepatic fat PRS adds prognostic information beyond traditional fibrosis scores (FIB-4), providing a more granular assessment of progression to severe liver disease and unmasking high-risk individuals within the clinically ambiguous intermediate-risk category who may benefit from intensified surveillance[108].
Personalized therapeutic strategies
Multi-layered evidence can be unified within a conceptual framework linking genotype, molecular phenotype, cellular and zonation phenotypes, clinical phenotype, and therapeutic targeting. PNPLA3 I148M illustrates this hierarchy: the risk allele (genotype) impairs triglyceride hydrolase activity and causes mutant protein accumulation on lipid droplets (molecular phenotype)[23], preferentially affecting periportal hepatocytes as demonstrated by single-cell eQTL mapping (cellular and zonation phenotype)[54], which manifests as accelerated MASH and fibrosis progression (clinical phenotype)[24], and is now being addressed by allele-specific siRNA and ASO therapeutics (therapeutic targeting)[93]. The same framework applies in reverse to protective alleles, such as HSD17B13 loss-of-function, where the therapeutic goal is the pharmacological phenocopy of a beneficial genotype[89]. In real-world clinical decision-making, these layers interact dynamically. Defining baseline susceptibility; epigenetic and environmental modifiers shape current disease activity; and the integration of multi-omics data with non-invasive fibrosis assessment guides treatment selection. For instance, a biopsy-proven MASH F2 patient who is a PNPLA3 G/G homozygote with concomitant obesity may be prioritized for combination therapy with metabolic agents (e.g., resmetirom, GLP-1 receptor agonists) and considered for enrollment in genotype-targeted siRNA/ASO trials.
Given MASLD’s complex, multifaceted pathophysiology, the transition from conventional “one-size-fits-all” treatment to precision medicine integrating patient-specific environmental, genetic, and epigenetic data is imperative. The convergence of genomic, transcriptomic, spatial multi-omics, and AI-based approaches now provides a multi-layered framework for this transition. Endotype-stratified approaches - using multi-omics integration with AI - have facilitated the identification of distinct molecular phenotypes (lipid-export failure, lipotoxic-inflammatory, fibrogenic, and cancer-prone trajectories) that conventional clinical phenotyping could not. Incorporating common risk alleles (PNPLA3, TM6SF2, GCKR, MBOAT7, HSD17B13, MARC1), DNA methylation modules, miRNA networks, and circulating protein/metabolite panels into unified risk engines might enable the prediction of rapid progression, extrahepatic complications, and pharmacologic responsiveness.
Genotype-guided therapeutic targeting
Human genetics has yielded the most robust evidence for precision medicine in MASLD. Several genetic variants - including PNPLA3 p.I148M, TM6SF2 p.E167K, MBOAT7, GCKR, HSD17B13, and MARC1 - are now established as major determinants of SLD susceptibility and progression[20,41]. PRS, a weighted sum of these risk alleles, enables the identification of individuals predisposed to severe disease and may be used for early risk stratification and prioritization of follow-up. The latest European Association for the Study of the Liver (EASL), European Association for the Study of Diabetes (EASD), and European Association for the Study of Obesity (EASO) guidelines recommend that clinicians in specialized centers may consider genetic risk profiling (e.g., PNPLA3 p.I148M and/or PRS) to personalize risk stratification, acknowledging that this concept merits further prospective validation[109].
A paradigmatic example of genotype-guided target discovery is the disease-specific eQTL approach, which identified AGXT2-rs2291702 as a genotype-dependent antifibrotic factor in MASLD[53]. AGXT2 overexpression attenuates fibrosis in mouse models, establishing a proof-of-concept that eQTL-driven target identification can yield therapeutically actionable candidates whose efficacy may be genotype-stratified. More recently, single-cell eQTL analysis revealed the FOXO1-rs13395911-EFHD1 axis as a novel genotype- and cell-state-dependent regulator of hepatocyte maladaptation in MASLD[54]. In rs13395911:TT carriers, progressive hepatocyte dysfunction results in the loss of EFHD1 expression, promoting ER stress, mitochondrial dysfunction, and apoptosis, suggesting that EFHD1-targeted interventions may be most effective in this specific genotypic population. These findings exemplify how genomic profiling can identify both the therapeutic target and the patient population that is most likely to benefit.
Nucleotide-based therapeutics and gene-specific interventions
The convergence of human genetic discoveries with nucleotide-based pharmacology has catalyzed genetically targeted therapies for MASLD[110]. The HSD17B13 rs72613567:TA loss-of-function variant, which confers protection against liver fibrosis, directly inspired the development of siRNA and ASO therapies aimed at silencing HSD17B13 expression[80,81]. These RNA-based therapeutics represent the first direct translation of MASLD genetics into pharmacological intervention. Similarly, active clinical development is underway for PNPLA3-targeting siRNA approaches, aiming to specifically reduce the toxic gain-of-function conferred by the PNPLA3 148M variant[76,77].
The sc-eQTL discovery that HSD17B13 regulation is zonation-dependent - with the strongest protective eQTL effect observed in periportal hepatocytes - has important implications for RNA therapeutic design[54]. If the pharmacological target gene is regulated in a zone-specific manner, understanding the spatial context of drug action is essential for optimizing therapeutic efficacy and minimizing off-target effects. Future spatial-eQTL studies mapping genotype-expression relationships across the hepatic lobule architecture are poised to further refine such spatially informed therapeutic strategies.
Genomics-informed drug repurposing represents another promising avenue. A recent cross-disciplinary analysis identified 212 putative causal genes for MASLD from large-scale multi-ancestry GWAS data, of which 57 encode druggable proteins, including targets of existing United States Food and Drug Administration (FDA)-approved drugs[111]. This approach may substantially accelerate therapeutic development by leveraging existing pharmacological knowledge for genetically validated targets.
Endotype-based patient stratification
Beyond single-gene stratification, data-driven clustering approaches are revealing clinically meaningful MASLD endotypes with distinct biological profiles and therapeutic implications. Jamialahmadi et al. applied partitioned PRS to identify two distinct types of MASLD associated with steatohepatitis: a “liver-specific” endotype, genetically linked and characterized by rapid progression of chronic liver disease, and a “cardiometabolic” endotype, primarily driven by dysglycemia and elevated triglycerides, associated with both chronic liver disease and elevated cardiovascular risk[52]. This endotype framework has been validated in independent Asian cohorts, supporting its cross-ethnic applicability[112].
Analogous subtype-specific insights have emerged from a multicenter, biopsy-proven SLD cohort study of 2,551 Korean individuals, classified as MASLD (n = 1,930), MetALD (n = 133), and ALD (n = 276) according to the new SLD nomenclature[20]. During a median follow-up of 43.8 months, MetALD [adjusted subdistribution hazard ratio (ASHR) 3.05; 95%CI: 1.08-8.58] and ALD (ASHR 5.80; 95%CI: 2.26-14.87) demonstrated substantially increased all-cause mortality risk compared with non-SLD. Liver-related event risks were even more pronounced: MetALD (ASHR 6.13; 95%CI: 1.33-28.20) and ALD (ASHR 10.96; 95%CI: 2.55-47.14). Critically, even within the advanced fibrosis (≥ F3) subgroup, ALD demonstrated higher mortality and liver-related event risks than MASLD, confirming the independent prognostic significance of the SLD subtype classification beyond fibrosis staging alone[20]. These findings reinforce the concept that tailored management strategies should account not only for fibrosis severity but also for the underlying etiology and molecular phenotype of SLD.
Spatial multi-omics and zonation-aware therapeutics
The liver lobule’s zonal architecture creates functionally distinct microenvironments that fundamentally shape disease biology. Recent advances in spatial transcriptomics and spatial metabolomics have provided unprecedented resolution of this heterogeneity. A landmark study generated single-cell and spatial transcriptomic and metabolomic maps from 61 human livers spanning control, isolated steatosis, and MASH stages. It identified microphthalmia-associated TF as a key regulator of lipid-associated macrophages (LAMs) and revealed a fibrosis-associated gene program enriched in advanced MASH - involving profibrotic RSPO3-LGR6 crosstalk between central vein endothelial cells and HSCs. Mass spectrometry imaging-based spatial metabolomics demonstrated MASLD-specific accumulation of phospholipids linked to LAM-mediated metabolism[113].
These spatial insights have direct therapeutic implications. If fibrogenic processes are driven by zonation-specific cellular crosstalk (e.g., RSPO3-LGR6 signaling in the pericentral region), therapeutic interventions targeting these pathways may need to be delivered or activated in a zone-specific manner. The effects of eQTLs vary across the portal-to-central vein axis - as shown by the periportal-specific regulation of HSD17B13 and CYP4V2 - underscore the need for spatially informed pharmacology[54]. Our ongoing spatial-eQTL projects, which aim to map genotype-expression relationships across 13 distinct lobular zones, will provide the comprehensive spatial regulatory atlas needed to guide such precision approaches.
From risk stratification to individualized treatment: an integrated framework
The convergence of these approaches points toward a multi-layered precision medicine framework for MASLD/SLD, as shown in Table 2.
Multi-layer framework for MASLD precision medicine (risk stratification to treatment selection)
| Stratification layer | Approach | Clinical application | Key evidence |
| Genetic risk | PRS, PNPLA3/HSD17B13 genotyping | Risk prediction, siRNA/ASO therapy selection | [109] |
| Disease-specific eQTL | Bulk and sc-eQTL mapping | Genotype-dependent target identification | [53,54] |
| Cell-state/zonation | sc-eQTL, spatial-eQTL | Zone-specific drug targeting | [54] |
| Clinical endotype | Partitioned PRS, data-driven clustering | Cardiometabolic vs. liver-specific pathway targeting | [52] |
| Multi-omics | Spatial transcriptomics + metabolomics | Target/biomarker discovery | [113] |
| AI-driven prediction | ML-based composite models | Treatment response prediction | [114] |
The first FDA-approved pharmacotherapy for MASLD (i.e., resmetirom) demonstrated that not all patients respond equally to treatment, with only approximately 30% achieving MASH resolution in pivotal trials[15]. This underscores the urgency of developing precision tools to identify responders a priori. By integrating genetic risk profiles, disease-specific eQTL data, spatial transcriptomic architecture, AI-based histologic assessment with digital pathology, and clinical endotyping, the field is moving toward a future in which therapeutic selection is individualized based on each patient’s unique molecular, genetic, and histologic phenotype.
Biomarker development and dynamic monitoring
Biomarker development must pivot from static, largely anatomical readouts toward dynamic, mechanistically anchored indicators directly linked to therapeutic targets. Genetic and epigenetic markers - such as ancestry-calibrated PRS, PNPLA3/TM6SF2/MBOAT7/HSD17B13/MARC1 combinations, complement-related methylation signatures, ATAC-seq-derived chromatin accessibility biomarkers (e.g., ETS family TF binding sites, ZNF384, IKZF1), DNMT1-dependent methylation modules, and miRNA panels - are attractive because they inform baseline risk, pathway dominance, and pharmacologic leverage points simultaneously[62,64].
FUTURE DIRECTIONS
Integration of multi-omics with AI
The advancement of MASLD precision medicine depends on integrating multi-omics data with advanced computational tools to resolve biologically meaningful endotypes invisible to routine clinical phenotyping. High-dimensional genomics, epigenomics, transcriptomics, proteomics, and metabolomics, when combined using AI and machine learning, can derive latent “molecular signatures” that distinguish disease trajectories rather than treating MASLD as a monolithic entity. For example, the integration of ATAC-seq chromatin accessibility data with DNA methylome profiling and RNA-seq transcriptomics - as demonstrated in Korean MASLD cohorts - enables identification of stage-specific regulatory elements and TF networks (e.g., HNF4α, PPARα, ETS family) that are epigenetically silenced during disease progression[62,64]. Similarly, combining genetic variant information (PNPLA3, TM6SF2, MARC1) with proteomic pathway analysis (EMT, hypoxia, EGFR signaling) provides mechanistic granularity for patient stratification beyond conventional histological scoring[97,98]. Such systems-biology frameworks incorporating common risk alleles, DNA methylation modules, miRNA networks, and circulating biomarkers into unified risk engines will enable prediction of rapid progression, extrahepatic complications, and pharmacologic responsiveness with unprecedented precision.
A critical frontier is the extension of eQTL mapping from bulk and single-cell levels to spatially resolved analyses. Our ongoing spatial-eQTL projects employing spatial transcriptomics on liver biopsies aim to systematically map genotype-expression associations across 13 distinct lobular zones, thereby identifying therapeutic targets whose regulatory activity is confined to specific microanatomical compartments. This spatial regulatory atlas, combined with organoid-derived liver-on-a-chip validation systems, is expected to provide mechanistic foundations for spatially targeted precision therapeutics[115]. Beyond research-stage multi-omics integration, AI is increasingly deployed in concrete clinical applications for MASLD. A landmark example is the AI-based digital pathology platform AIM-MASH, which has demonstrated superior performance over unassisted pathologist reads for inflammation, ballooning, and MASH resolution scoring in regulatory-grade validation studies and was recently qualified by the U.S. FDA as the first AI-based drug development tool to support histologic endpoint assessment in MASH clinical trials[116]. AI-augmented magnetic resonance imaging-derived proton density fat fraction (MRI-PDFF) and elastography are also expanding the toolkit for non-invasive longitudinal monitoring of hepatic steatosis and fibrosis, complementing biopsy-based assessment[117]. Multimodal AI models that jointly integrate genomic, transcriptomic, histological, and imaging data have emerged as the foundation for automated endotype classification and individualized treatment selection in routine hepatology practice[114].
Clinical trial design evolution
Clinical trial design for MASLD must shift from “all-comers” recruitment to genetically and endotype-enriched cohorts. Up-front stratification with PRS, key single-gene variants, and body-composition indices (including sarcopenia assessment) enables more efficient trial conduct. Adaptive platform trials incorporating real-time biomarker feedback could dynamically reassign non-responders to alternative mechanism-based arms, reduce the required sample sizes for meaningful effect detection, and accelerate combination regimen decision-making. Embedding multi-omics profiling and dynamic biomarkers into trial eligibility, stratification frameworks, and endpoints will be critical for translating proof-of-concept nucleic-acid and epigenetic therapies into scalable, personalized treatment algorithms implementable in routine MASLD care.
Precision prevention and early interception
Beyond therapeutic development, precision medicine enables earlier disease interception through risk stratification of asymptomatic individuals. Population-based genetic screening using PRS can identify high-risk individuals for targeted imaging surveillance and preventive interventions (lifestyle modification, pharmaceutical prevention) before the development of advanced fibrosis, thus fundamentally shifting MASLD management from reactive to proactive paradigms.
BARRIERS TO CLINICAL IMPLEMENTATION AND TRANSLATIONAL ROADMAP
Despite rapid advances in MASLD genetics, epigenetics, and multi-omics, several practical barriers must be addressed before precision medicine can be incorporated into routine hepatology practice.
Cost and accessibility
Whole-genome sequencing, snRNA-seq, and spatial transcriptomics remain confined to research-intensive centers because of their high cost and infrastructure requirements. For routine clinical use, targeted genotyping panels covering the major risk and protective variants (PNPLA3, TM6SF2, GCKR, MBOAT7, HSD17B13, MARC1) offer a pragmatic and substantially less costly alternative, capturing the majority of clinically actionable common variants while minimizing data management and incidental-finding
PRS standardization and cross-ancestry portability
Most current MASLD PRS are derived from cohorts of European ancestry and demonstrate reduced predictive performance in non-European populations, with the greatest loss of accuracy observed in individuals of African ancestry[118]. Methodological advances such as multi-ancestry meta-analyses and Bayesian frameworks leveraging shared cross-ancestry effects have improved portability[119]. Ancestry-diverse GWAS reference datasets and standardized cross-population PRS validation will be necessary for equitable clinical implementation.
Regulatory and ethical considerations
Clinical implementation of genetic testing for MASLD requires distinct considerations[120]. Although the major MASLD risk variants are common polymorphisms with modest individual effect sizes, appropriate informed consent and genetic counseling infrastructure remain essential, particularly when testing is a part of broader genomic panel that may yield incidental findings. Protection against genetic discrimination varies substantially across jurisdictions and many countries, including those in East Asia, lack comprehensive legislation. Moreover, the regulatory pathways of the PRS-based clinical decision-making tools remain unclear.
Integration into routine clinical workflows
Effective use of genetic and biomarker data in routine practice requires (i) electronic health record-embedded clinical decision support that translates genotype, PRS, and non-invasive fibrosis scores into actionable risk stratification at the point of care; (ii) structured education to enhance the genomic literacy of hepatologists and primary care physicians; and (iii) multidisciplinary care models combining hepatology, endocrinology, and clinical genetics expertise. The 2024 EASL-EASD-EASO guidelines provide an initial clinical entry point by endorsing referral for genetic evaluation in patients with a strong family history of severe liver disease or early severe phenotypes[109].
CONCLUSION
In summary, MASLD reflects a multi-layered interaction between inherited susceptibility, metabolic exposures, and epigenetic regulation, which together drive heterogeneity in fibrosis progression and HCC risk. Advances in genetic discovery and polygenic risk scoring are beginning to enable clinically meaningful risk stratification, particularly when calibrated for ancestry and combined with non-invasive biomarkers. Functional genomics, single-cell and spatial multi-omics, and epigenetic profiling are clarifying cell-type- and context-specific mechanisms that link genotype to phenotype and highlight actionable pathways. These insights support a shift toward biomarker- and mechanism-informed trial design and, ultimately, individualized prevention and therapy - ranging from liver-directed nucleic-acid therapeutics to emerging epigenetic and metabolic interventions. Key next steps include prospective validation in diverse populations, harmonized reporting of effect sizes and uncertainty, and pragmatic implementation studies addressing cost, equity, and clinical workflows. Progress on these fronts will be essential to translate precision medicine from discovery to routine care for MASLD and its complications.
DECLARATIONS
Acknowledgments
We thank MedVisual for creating Figure 1 and the Graphical Abstract.
Authors’ contributions
Manuscript conceptualization, literature review, initial draft preparation, and critical revision for intellectual content: Choi E, Lee YS
Literature review and critical revision for important intellectual content: Lee DH
Study conceptualization, manuscript supervision, critical revision for important intellectual content, and final approval of the version to be published: Kim W
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
During the preparation of this manuscript, the AI tool Google Gemini (version 3 Pro, released 2025-11-18) was used solely for language editing and improvement of English expression. The tool did not influence the study design, data collection, analysis, interpretation, or the scientific content of the work. All authors take full responsibility for the accuracy, integrity, and final content of the manuscript.
Financial support and sponsorship
This work is supported by grants from the National Research Foundation (NRF) of Korea (RS-2021-NR056442, RS-2021-NR061523, RS-2022-NR067269, RS-202300223831, RS-2024-00439963, RS-2024-00440883, and RS-2025-25458964).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Younossi ZM, Kalligeros M, Henry L. Epidemiology of metabolic dysfunction-associated steatotic liver disease. Clin Mol Hepatol. 2025;31:S32-50.
2. Younossi ZM, Stepanova M, Al Shabeeb R, et al. The changing epidemiology of adult liver transplantation in the United States in 2013-2022: the dominance of metabolic dysfunction-associated steatotic liver disease and alcohol-associated liver disease. Hepatol Commun. 2024;8:e0352.
3. Rinella ME, Lazarus JV, Ratziu V, et al.; NAFLD Nomenclature consensus group. A multisociety Delphi consensus statement on new fatty liver disease nomenclature. J Hepatol. 2023;79:1542-56.
4. Sohn W, Lee YS, Kim SS, et al.; Korean Association for the Study of the Liver (KASL). KASL clinical practice guidelines for the management of metabolic dysfunction-associated steatotic liver disease 2025. Clin Mol Hepatol. 2025;31:S1-31.
5. Younossi ZM, Golabi P, Paik J, et al. Prevalence of metabolic dysfunction-associated steatotic liver disease in the Middle East and North Africa. Liver Int. 2024;44:1061-70.
6. Lekakis V, Papatheodoridis GV. Natural history of metabolic dysfunction-associated steatotic liver disease. Eur J Intern Med. 2024;122:3-10.
7. Sakai K, Shima T, Oya H, et al. Association between liver fibrosis and cause-specific mortality in japanese patients with biopsy-confirmed metabolic dysfunction-associated steatotic liver disease: a prospective cohort study/liver fibrosis and mortality in Japanese MASLD. Hepatol Res. 2026;56:33-49.
8. Miao L, Targher G, Byrne CD, Cao YY, Zheng MH. Current status and future trends of the global burden of MASLD. Trends Endocrinol Metab. 2024;35:697-707.
9. Charlton MR, Burns JM, Pedersen RA, Watt KD, Heimbach JK, Dierkhising RA. Frequency and outcomes of liver transplantation for nonalcoholic steatohepatitis in the United States. Gastroenterology. 2011;141:1249-53.
10. Sanna C, Rosso C, Marietti M, Bugianesi E. Non-alcoholic fatty liver disease and extra-hepatic cancers. Int J Mol Sci. 2016;17:717.
11. Li AA, Ahmed A, Kim D. Extrahepatic manifestations of nonalcoholic fatty liver disease. Gut Liver. 2020;14:168-78.
12. Bansal SK, Bansal MB. Pathogenesis of MASLD and MASH - role of insulin resistance and lipotoxicity. Aliment Pharmacol Ther. 2024;59:S10-22.
13. Termite F, Archilei S, D’Ambrosio F, et al. Gut microbiota at the crossroad of hepatic oxidative stress and MASLD. Antioxidants. 2025;14:56.
14. DiStefano JK, Gerhard GS. A complement to epigenetics in metabolic dysfunction-associated steatotic liver disease: Editorial on “DNA methylome analysis reveals epigenetic alteration of complement genes in advanced metabolic dysfunction-associated steatotic liver disease”. Clin Mol Hepatol. 2025;31:297-300.
15. Harrison SA, Bedossa P, Guy CD, et al.; MAESTRO-NASH Investigators. A phase 3, randomized, controlled trial of resmetirom in NASH with liver fibrosis. N Engl J Med. 2024;390:497-509.
16. Sanyal AJ, Newsome PN, Kliers I, et al.; ESSENCE Study Group. Phase 3 trial of semaglutide in metabolic dysfunction-associated steatohepatitis. N Engl J Med. 2025;392:2089-99.
17. Ren Q, Zhi L, Liu H. Semaglutide therapy and accelerated sarcopenia in older adults with type 2 diabetes: a 24-month retrospective cohort study. Drug Des Devel Ther. 2025;19:5645-52.
18. Ayesh H, Beran A, Suhail S, Ayesh S, Niswender K. Efficacy and safety of resmetirom in MASLD and MASH: network meta-analysis of randomized clinical trials. J Basic Clin Physiol Pharmacol. 2025;36:3-11.
19. Huang X, Wu M, Huang B, Zhang Y. Gastrointestinal adverse events associated with GLP-1 receptor agonists in metabolic dysfunction-associated steatotic liver disease (MASLD): a systematic review and meta-analysis. Front Med. 2025;12:1509947.
20. Kim GA, Jang H, Kim MY, et al. Multicenter prospective cohort study on clinical outcomes and fibrosis patterns in biopsy-proven steatotic liver disease subtypes. Gastroenterology. 2026;170:1571-83.
21. Cirulli ET, Goldstein DB. Uncovering the roles of rare variants in common disease through whole-genome sequencing. Nat Rev Genet. 2010;11:415-25.
22. Haas ME, Pirruccello JP, Friedman SN, et al. Machine learning enables new insights into genetic contributions to liver fat accumulation. Cell Genom. 2021;1:100066.
23. BasuRay S, Wang Y, Smagris E, Cohen JC, Hobbs HH. Accumulation of PNPLA3 on lipid droplets is the basis of associated hepatic steatosis. Proc Natl Acad Sci U S A. 2019;116:9521-6.
24. Romeo S, Kozlitina J, Xing C, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2008;40:1461-5.
25. Kozlitina J, Sookoian S. Global epidemiological impact of PNPLA3 I148M on liver disease. Liver Int. 2025;45:e16123.
26. Mahdessian H, Taxiarchis A, Popov S, et al. TM6SF2 is a regulator of liver fat metabolism influencing triglyceride secretion and hepatic lipid droplet content. Proc Natl Acad Sci U S A. 2014;111:8913-8.
27. Liu YL, Reeves HL, Burt AD, et al. TM6SF2 rs58542926 influences hepatic fibrosis progression in patients with non-alcoholic fatty liver disease. Nat Commun. 2014;5:4309.
28. Li XY, Liu Z, Li L, Wang HJ, Wang H. TM6SF2 rs58542926 is related to hepatic steatosis, fibrosis and serum lipids both in adults and children: a meta-analysis. Front Endocrinol. 2022;13:1026901.
29. Kozlitina J, Smagris E, Stender S, et al. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2014;46:352-6.
30. Beer NL, Tribble ND, McCulloch LJ, et al. The P446L variant in GCKR associated with fasting plasma glucose and triglyceride levels exerts its effect through increased glucokinase activity in liver. Hum Mol Genet. 2009;18:4081-8.
31. Santoro N, Zhang CK, Zhao H, et al. Variant in the glucokinase regulatory protein (GCKR) gene is associated with fatty liver in obese children and adolescents. Hepatology. 2012;55:781-9.
32. Caddeo A, Jamialahmadi O, Solinas G, et al. MBOAT7 is anchored to endomembranes by six transmembrane domains. J Struct Biol. 2019;206:349-60.
33. Thangapandi VR, Knittelfelder O, Brosch M, et al. Loss of hepatic Mboat7 leads to liver fibrosis. Gut. 2021;70:940-50.
34. Thabet K, Asimakopoulos A, Shojaei M, et al.; International Liver Disease Genetics Consortium. MBOAT7 rs641738 increases risk of liver inflammation and transition to fibrosis in chronic hepatitis C. Nat Commun. 2016;7:12757.
35. Teo K, Abeysekera KWM, Adams L, et al.; EU-PNAFLD Investigators, GOLD Consortium. rs641738C>T near MBOAT7 is associated with liver fat, ALT and fibrosis in NAFLD: a meta-analysis. J Hepatol. 2021;74:20-30.
36. Liu S, Gao Y, Zhang C, et al. SAMM50 affects mitochondrial morphology through the association of Drp1 in mammalian cells. FEBS Lett. 2016;590:1313-23.
37. Li Z, Shen W, Wu G, et al. The role of SAMM50 in non-alcoholic fatty liver disease: from genetics to mechanisms. FEBS Open Bio. 2021;11:1893-906.
38. Park JH, Park KJ. Genetic variants associated with metabolic dysfunction-associated fatty liver diseases in a Korean population. Eur J Med Res. 2025;30:318.
39. Hsu SW, Lin MR, Chou WH, Wan YY, Kao WY, Chang WC. Cross-ancestry discovery of genetic risk variants for lean metabolic dysfunction-associated steatotic liver disease. Cell Biosci. 2025;15:131.
40. Chung GE, Lee Y, Yim JY, et al. Genetic polymorphisms of PNPLA3 and SAMM50 are associated with nonalcoholic fatty liver disease in a Korean population. Gut Liver. 2018;12:316-23.
41. Demirtas CO, Yilmaz Y. Decoding 17-beta-hydroxysteroid dehydrogenase 13: a multifaceted perspective on its role in hepatic steatosis and associated disorders. J Clin Transl Hepatol. 2024;12:857-64.
42. Su W, Wu S, Yang Y, et al. Phosphorylation of 17β-hydroxysteroid dehydrogenase 13 at serine 33 attenuates nonalcoholic fatty liver disease in mice. Nat Commun. 2022;13:6577.
43. Abul-Husn NS, Cheng X, Li AH, et al. A protein-truncating HSD17B13 variant and protection from chronic liver disease. N Engl J Med. 2018;378:1096-106.
44. Luukkonen PK, Tukiainen T, Juuti A, et al. Hydroxysteroid 17-β dehydrogenase 13 variant increases phospholipids and protects against fibrosis in nonalcoholic fatty liver disease. JCI Insight. 2020;5:132158.
45. Dutta T, Sasidharan K, Ciociola E, et al. Mitochondrial amidoxime-reducing component 1 p.Ala165Thr increases protein degradation mediated by the proteasome. Liver Int. 2024;44:1219-32.
46. Emdin CA, Haas ME, Khera AV, et al.; Million Veteran Program. A missense variant in Mitochondrial Amidoxime Reducing Component 1 gene and protection against liver disease. PLoS Genet. 2020;16:e1008629.
47. Koo BK, Joo SK, Kim D, et al. Additive effects of PNPLA3 and TM6SF2 on the histological severity of non-alcoholic fatty liver disease. J Gastroenterol Hepatol. 2018;33:1277-85.
48. Sun B, Ding X, Tan J, et al. TM6SF2 E167K variant decreases PNPLA3-mediated PUFA transfer to promote hepatic steatosis and injury in MASLD. Clin Mol Hepatol. 2024;30:863-82.
49. Paternostro R, Staufer K, Traussnigg S, et al. Combined effects of PNPLA3, TM6SF2 and HSD17B13 variants on severity of biopsy-proven non-alcoholic fatty liver disease. Hepatol Int. 2021;15:922-33.
50. Gao F, Zheng KI, Chen SD, et al. Individualized polygenic risk score identifies NASH in the Eastern Asia region: a derivation and validation study. Clin Transl Gastroenterol. 2021;12:e00321.
51. Koo BK, Joo SK, Kim D, et al. Development and validation of a scoring system, based on genetic and clinical factors, to determine risk of steatohepatitis in Asian patients with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol. 2020;18:2592-9.e10.
52. Jamialahmadi O, De Vincentis A, Tavaglione F, et al. Partitioned polygenic risk scores identify distinct types of metabolic dysfunction-associated steatotic liver disease. Nat Med. 2024;30:3614-23.
53. Yoo T, Joo SK, Kim HJ, et al.; Innovative Target Exploration of NAFLD (ITEN) consortium. Disease-specific eQTL screening reveals an anti-fibrotic effect of AGXT2 in non-alcoholic fatty liver disease. J Hepatol. 2021;75:514-23.
54. Hong SE, Mun SJ, Lee YJ, et al. Single-cell eQTL analysis identifies genetic variation underlying metabolic dysfunction-associated steatohepatitis. Nat Genet. 2025;57:1638-48.
55. Xu X, Xu H, Liu X, et al. MBOAT7 rs641738 (C>T) is associated with NAFLD progression in men and decreased ASCVD risk in elder Chinese population. Front Endocrinol. 2023;14:1199429.
56. Young S, Tariq R, Provenza J, et al. Prevalence and profile of nonalcoholic fatty liver disease in lean adults: systematic review and meta-analysis. Hepatol Commun. 2020;4:953-72.
57. Lee Y, Cho EJ, Choe EK, et al. Genome-wide association study of metabolic dysfunction-associated fatty liver disease in a Korean population. Sci Rep. 2024;14:9753.
58. Lee J, Cha JH, Cho HS, et al. The PNPLA3 I148M variant is associated with immune cell infiltration and advanced fibrosis in MASLD: a prospective genotype-phenotype study. J Gastroenterol. 2025;60:1284-95.
59. Ko YK, Kim H, Lee Y, Lee YS, Gim JA. DNA methylation patterns according to fatty liver index and longitudinal changes from the Korean Genome and Epidemiology Study (KoGES). Curr Issues Mol Biol. 2022;44:1149-68.
60. Magdy A, Kim HJ, Go H, et al. DNA methylome analysis reveals epigenetic alteration of complement genes in advanced metabolic dysfunction-associated steatotic liver disease. Clin Mol Hepatol. 2024;30:824-44.
61. Ahn J, Kim S, Jeong JY, et al. Whole genome DNA methylation patterns in tissue and cfDNA associated with fibrosis reflect the complex signature of MASLD. PLoS One. 2025;20:e0328207.
62. Sohn HA, Go H, An TH, et al. DNMT1 facilitates the progression of MASLD by impeding transcription mediated by HNF4α and PPARα. Clin Mol Hepatol. 2026;Epub ahead of print.
63. Claveria-Cabello A, Colyn L, Arechederra M, et al. Epigenetics in liver fibrosis: could HDACs be a therapeutic target? Cells. 2020;9:2321.
64. Kang B, Kang B, Roh TY, Seong RH, Kim W. The chromatin accessibility landscape of nonalcoholic fatty liver disease progression. Mol Cells. 2022;45:343-52.
66. Liu CH, Ampuero J, Gil-Gómez A, et al. miRNAs in patients with non-alcoholic fatty liver disease: a systematic review and meta-analysis. J Hepatol. 2018;69:1335-48.
67. Liu XL, Pan Q, Cao HX, et al. Lipotoxic hepatocyte-derived exosomal microRNA 192-5p activates macrophages through Rictor/Akt/Forkhead box transcription factor O1 signaling in nonalcoholic fatty liver disease. Hepatology. 2020;72:454-69.
68. Tobaruela-Resola AL, Milagro FI, Mogna-Pelaez P, Moreno-Aliaga MJ, Abete I, Zulet MÁ. The use of circulating miRNAs for the diagnosis, prognosis, and personalized treatment of MASLD. J Physiol Biochem. 2025;81:589-609.
69. Kim TH, Lee Y, Lee YS, et al. Circulating miRNA is a useful diagnostic biomarker for nonalcoholic steatohepatitis in nonalcoholic fatty liver disease. Sci Rep. 2021;11:14639.
70. Carpi S, Daniele S, de Almeida JFM, Gabbia D. Recent advances in miRNA-based therapy for MASLD/MASH and MASH-associated HCC. Int J Mol Sci. 2024;25:12229.
71. Kim D, Kim W, Joo SK, et al. Association between body size-metabolic phenotype and nonalcoholic steatohepatitis and significant fibrosis. J Gastroenterol. 2020;55:330-41.
72. Son H, Koo BK, Joo SK, et al.; Innovative Target Exploration of NAFLD (ITEN) consortium. PNPLA3 genotypes modify the adverse effect of the total energy intake on high-risk nonalcoholic steatohepatitis development. Am J Clin Nutr. 2023;117:910-7.
73. Smith GI, Shankaran M, Yoshino M, et al. Insulin resistance drives hepatic de novo lipogenesis in nonalcoholic fatty liver disease. J Clin Invest. 2020;130:1453-60.
74. Chiu S, Mulligan K, Schwarz JM. Dietary carbohydrates and fatty liver disease: de novo lipogenesis. Curr Opin Clin Nutr Metab Care. 2018;21:277-82.
75. Geidl-Flueck B, Gerber PA. Fructose drives de novo lipogenesis affecting metabolic health. J Endocrinol. 2023;257:e220270.
76. Petersen MC, Shulman GI. Roles of diacylglycerols and ceramides in hepatic insulin resistance. Trends Pharmacol Sci. 2017;38:649-65.
77. Galbo T, Perry RJ, Jurczak MJ, et al. Saturated and unsaturated fat induce hepatic insulin resistance independently of TLR-4 signaling and ceramide synthesis in vivo. Proc Natl Acad Sci U S A. 2013;110:12780-5.
78. Mirza MS. Obesity, visceral Fat, and NAFLD: querying the role of adipokines in the progression of nonalcoholic fatty liver disease. ISRN Gastroenterol. 2011;2011:592404.
79. Schrager MA, Metter EJ, Simonsick E, et al. Sarcopenic obesity and inflammation in the InCHIANTI study. J Appl Physiol. 2007;102:919-25.
80. Song W, Yoo SH, Jang J, et al. Association between sarcopenic obesity status and nonalcoholic fatty liver disease and fibrosis. Gut Liver. 2023;17:130-8.
81. Agbaje AO. Accelerometer-based sedentary time and physical activity with MASLD and liver cirrhosis in 2684 British adolescents. npj Gut Liver. 2024;1:2.
82. Wei X, Liu X, Zhao J, Zhang Y, Qiu L, Zhang J. Association of sarcopenia and physical activity on the severity of metabolic dysfunction-associated steatotic liver disease among United States adults: NHANES 2017 - 2018. Front Aging. 2025;6:1573170.
83. Nobili V, Liccardo D, Bedogni G, et al. Influence of dietary pattern, physical activity, and I148M PNPLA3 on steatosis severity in at-risk adolescents. Genes Nutr. 2014;9:392.
84. Nishioji K, Mochizuki N, Kobayashi M, et al. The impact of PNPLA3 rs738409 genetic polymorphism and weight gain ≥ 10 kg after age 20 on non-alcoholic fatty liver disease in non-obese japanese individuals. PLoS One. 2015;10:e0140427.
85. O’Hare EA, Yang R, Yerges-Armstrong LM, et al. TM6SF2 rs58542926 impacts lipid processing in liver and small intestine. Hepatology. 2017;65:1526-42.
86. Meroni M, Dongiovanni P, Longo M, et al. Mboat7 down-regulation by hyper-insulinemia induces fat accumulation in hepatocytes. EBioMedicine. 2020;52:102658.
87. Tanaka Y, Shimanaka Y, Caddeo A, et al. LPIAT1/MBOAT7 depletion increases triglyceride synthesis fueled by high phosphatidylinositol turnover. Gut. 2021;70:180-93.
88. Fabbrini E, Rady B, Koshkina A, et al. Phase 1 trials of PNPLA3 siRNA in I148M homozygous patients with MAFLD. N Engl J Med. 2024;391:475-6.
89. Sanyal AJ, Taubel J, Badri P, et al. Phase I randomized double-blind study of an RNA interference therapeutic targeting HSD17B13 for metabolic dysfunction-associated steatohepatitis. J Hepatol. 2025;83:838-48.
90. Mak LY, Gane E, Schwabe C, et al. A phase I/II study of ARO-HSD, an RNA interference therapeutic, for the treatment of non-alcoholic steatohepatitis. J Hepatol. 2023;78:684-92.
91. Armisen J, Rauschecker M, Sarv J, et al. AZD2693, a PNPLA3 antisense oligonucleotide, for the treatment of MASH in 148M homozygous participants: two randomized phase I trials. J Hepatol. 2025;83:31-42.
92. Li S, Xiong F, Zhang S, et al. Oligonucleotide therapies for nonalcoholic steatohepatitis. Mol Ther Nucleic Acids. 2024;35:102184.
93. Murray JK, Long J, Liu L, et al. Identification and optimization of a minor allele-specific siRNA to prevent PNPLA3 I148M-driven nonalcoholic fatty liver disease. Nucleic Acid Ther. 2021;31:324-40.
94. Eli Lilly and Company. A single-ascending and repeated dose study of ly3849891 in participants with metabolic dysfunction-associated steatotic liver disease (MASLD). Available from: https://trials.lilly.com/en-US/trial/345456. [Last accessed on 18 Jun 2026].
95. ClinicalTrialsgov. A study to learn about the study medicine PF-07853578 and how it acts in the bodies of healthy adults. Available from: https://clinicaltrials.gov/study/NCT05890105. [Last accessed on 18 Jun 2026].
96. Lindén D, Ahnmark A, Pingitore P, et al. Pnpla3 silencing with antisense oligonucleotides ameliorates nonalcoholic steatohepatitis and fibrosis in Pnpla3 I148M knock-in mice. Mol Metab. 2019;22:49-61.
97. Ciociola E, Dutta T, Sasidharan K, et al. Downregulation of the MARC1 p.A165 risk allele reduces hepatocyte lipid content by increasing beta-oxidation. Clin Mol Hepatol. 2025;31:445-59.
98. Kovooru L, Zhang J, Monni FG, et al. MTARC1 p.A165 ablation reduces hepatocellular carcinoma aggressiveness in vitro and in vivo. Clin Mol Hepatol. 2026;32:829-42.
99. Rodríguez-Sanabria JS, Escutia-Gutiérrez R, Rosas-Campos R, Armendáriz-Borunda JS, Sandoval-Rodríguez A. An update in epigenetics in metabolic-associated fatty liver disease. Front Med. 2021;8:770504.
100. Van Dijck E, Van Laere S, Logie E, et al. Gradual DNA methylation changes reveal transcription factors implicated in metabolic dysfunction-associated steatotic liver disease progression and epigenetic age acceleration. Clin Epigenetics. 2025;17:138.
101. Pant R, Kabeer SW, Sharma S, et al. Pharmacological inhibition of DNMT1 restores macrophage autophagy and M2 polarization in Western diet-induced nonalcoholic fatty liver disease. J Biol Chem. 2023;299:104779.
102. Tapadar S, Fathi S, Wu B, et al. Liver-targeting class I selective histone deacetylase inhibitors potently suppress hepatocellular tumor growth as standalone agents. Cancers. 2020;12:3095.
103. Mannaerts I, Eysackers N, Onyema OO, et al. Class II HDAC inhibition hampers hepatic stellate cell activation by induction of microRNA-29. PLoS One. 2013;8:e55786.
104. Liu AM, Xu Z, Shek FH, et al. miR-122 targets pyruvate kinase M2 and affects metabolism of hepatocellular carcinoma. PLoS One. 2014;9:e86872.
105. Singh R, Ha SE, Wei L, et al. miR-10b-5p rescues diabetes and gastrointestinal dysmotility. Gastroenterology. 2021;160:1662-78.e18.
106. Hanin G, Yayon N, Tzur Y, et al. miRNA-132 induces hepatic steatosis and hyperlipidaemia by synergistic multitarget suppression. Gut. 2018;67:1124-34.
107. Trajkovski M, Hausser J, Soutschek J, et al. MicroRNAs 103 and 107 regulate insulin sensitivity. Nature. 2011;474:649-53.
108. De Vincentis A, Tavaglione F, Jamialahmadi O, et al. A polygenic risk score to refine risk stratification and prediction for severe liver disease by clinical fibrosis scores. Clin Gastroenterol Hepatol. 2022;20:658-73.
109. European Association for the Study of the Liver (EASL), European Association for the Study of Diabetes (EASD), European Association for the Study of Obesity (EASO). EASL-EASD-EASO Clinical Practice Guidelines on the management of metabolic dysfunction-associated steatotic liver disease (MASLD). J Hepatol. 2024;81:492-542.
110. Caddeo A, Romeo S. Precision medicine and nucleotide-based therapeutics to treat steatotic liver disease. Clin Mol Hepatol. 2025;31:S76-93.
111. Seagle HM, Akerele AT, DeCorte JA, et al. Genomics-informed drug repurposing strategy identifies novel therapeutic targets for metabolic dysfunction-associated steatotic liver disease. medRxiv 2025;2025.02.18.25321035. Available from: https://www.medrxiv.org/content/10.1101/2025.02.18.25321035v1. [accessed 18 June 2026].
112. Zhou XD, Song SJ, Guo CY, et al. Validation of a data-driven clustering model for MASLD: evidence from three large-scale Asian cohorts. JHEP Rep. 2026;8:101645.
113. Li Z, Luo G, Gan C, et al. Spatially resolved multi-omics of human metabolic dysfunction-associated steatotic liver disease. Nat Genet. 2025;57:3112-25.
114. Thakral N, Desalegn H, Diaz LA, et al. A precision medicine guided approach to the utilization of biomarkers in MASLD. Semin Liver Dis. 2024;44:273-86.
115. Yuan L, Dawka S, Kim Y, et al. Human assembloids recapitulate periportal liver tissue in vitro. Nature. 2026;650:438-49.
116. Pulaski H, Harrison SA, Mehta SS, et al. Clinical validation of an AI-based pathology tool for scoring of metabolic dysfunction-associated steatohepatitis. Nat Med. 2025;31:315-22.
117. Maghsoudi H, Khonche A, Gereami R, Gharebakhshi F. Physics-aware imaging AI for quantitative MASLD biomarker mapping: a systematic review of deep learning and radiomics across ultrasound, CT, and MRI. Abdom Radiol. 2026;51:2919-31.
118. Kachuri L, Chatterjee N, Hirbo J, et al.; Polygenic Risk Methods in Diverse Populations (PRIMED) Consortium Methods Working Group. Principles and methods for transferring polygenic risk scores across global populations. Nat Rev Genet. 2024;25:8-25.
119. Hoggart CJ, Choi SW, García-González J, Souaiaia T, Preuss M, O’Reilly PF. BridgePRS leverages shared genetic effects across ancestries to increase polygenic risk score portability. Nat Genet. 2024;56:180-6.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Special Topic
Copyright

Data & Comments
Data
0












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].