


Immune escape as an ecosystem state in cancer
 0
0 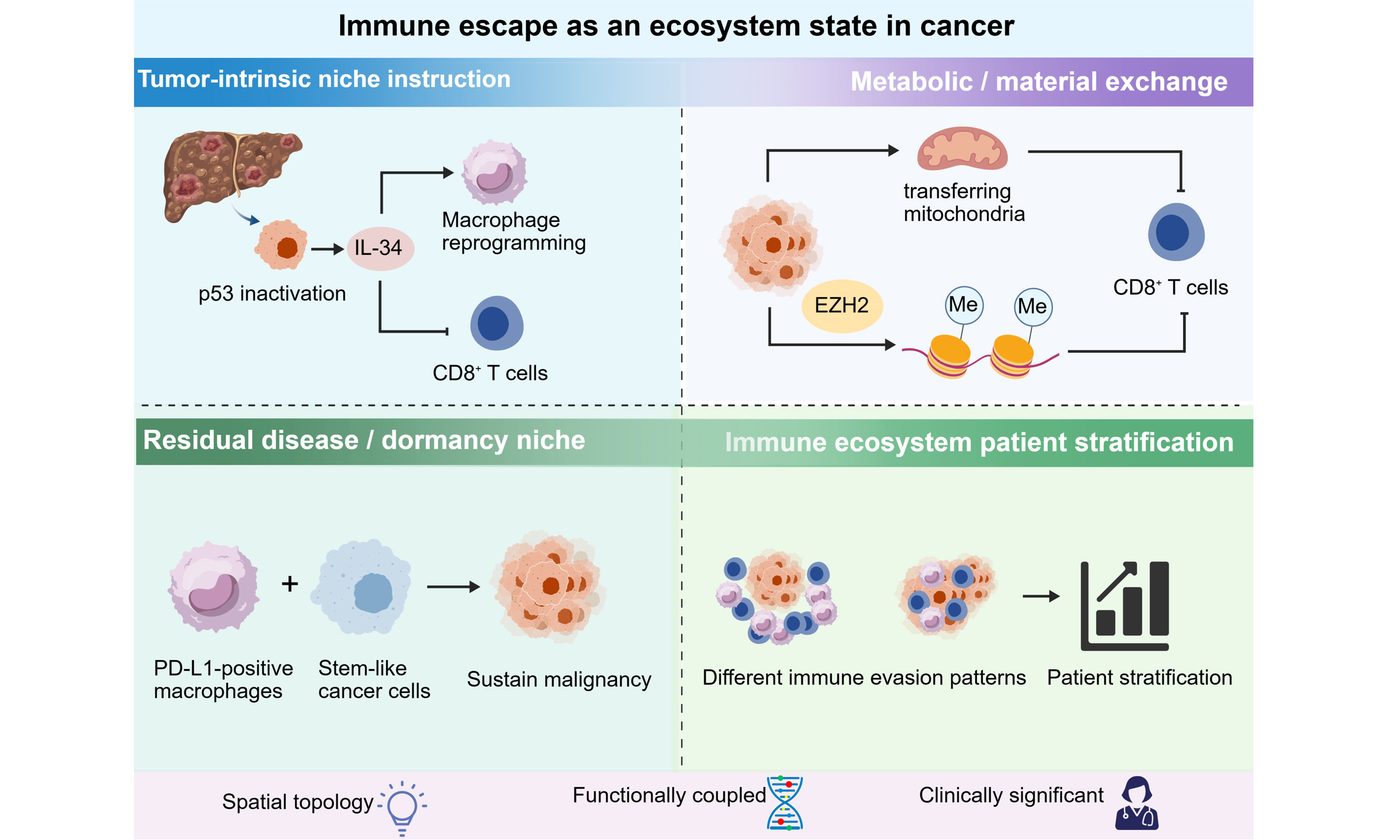
INTRODUCTION
The tumor immune microenvironment (TIME) still largely relies on compositional profiling. Tumors are classified into inflamed, immune-excluded, and immune-desert subtypes, and immune activity is often inferred from the abundance of lymphocytes, macrophages, fibroblasts, or specific inhibitory ligands. Those descriptors remain useful, but they are no longer sufficient. They encompass broad phenotypes while leaving a more difficult set of questions unresolved: why tumors with similar immune composition behave differently, why they exhibit varied responses to checkpoint blockade, and why relapse can occur through markedly different pathways[1,2].
Recent advances in tumor immunology and spatial oncology have rendered this limitation increasingly difficult to ignore. The primary issue in many tumors is not a lack of immune cells, but rather that these cells localize to unfavorable niches, interact with unsuitable neighboring cells, or function under local conditions that compromise their effectiveness. Spatial transcriptomics, multiplex imaging, and related approaches have shown that the importance of a specific immune or stromal population is highly dependent on tissue architecture, neighborhood organization, and the physical accessibility of malignant targets[3,4]. A CD8+-enriched infiltrate may remain functionally uninformative if confined to stromal compartments, whereas macrophages bearing identical surface markers can execute vastly divergent transcriptional programs depending on their association with vasculature, fibroblasts, damaged tissue, or stem-like malignant cells[3,4]. Translational reviews of spatial biomarkers have consistently emphasized from a clinical perspective, that these assays are particularly promising because they preserve the architecture that conventional bulk or low-plex approaches tend to obscure[5,6].
This perspective argues that the clinically relevant unit of immune failure lies not in composition alone, but the immune-escape ecosystem: a spatially organized, functionally interconnected, and clinically significant tissue state where malignant cells, stromal and vascular components, immune populations, and regional metabolic conditions converge to impair immune surveillance and sustain tumor viability. This framework is useful not because it complicates the research field, but because it provides a more effective explanation for several enduring problems: why abundance-based biomarkers often prove disappointing, why mechanistic findings that appear convincing in one context fail to generalize effectively in another, and why enhanced descriptive resolution has not yet resulted in correspondingly robust clinical tools[1,3,5]. A schematic overview of this framework is shown in Figure 1.
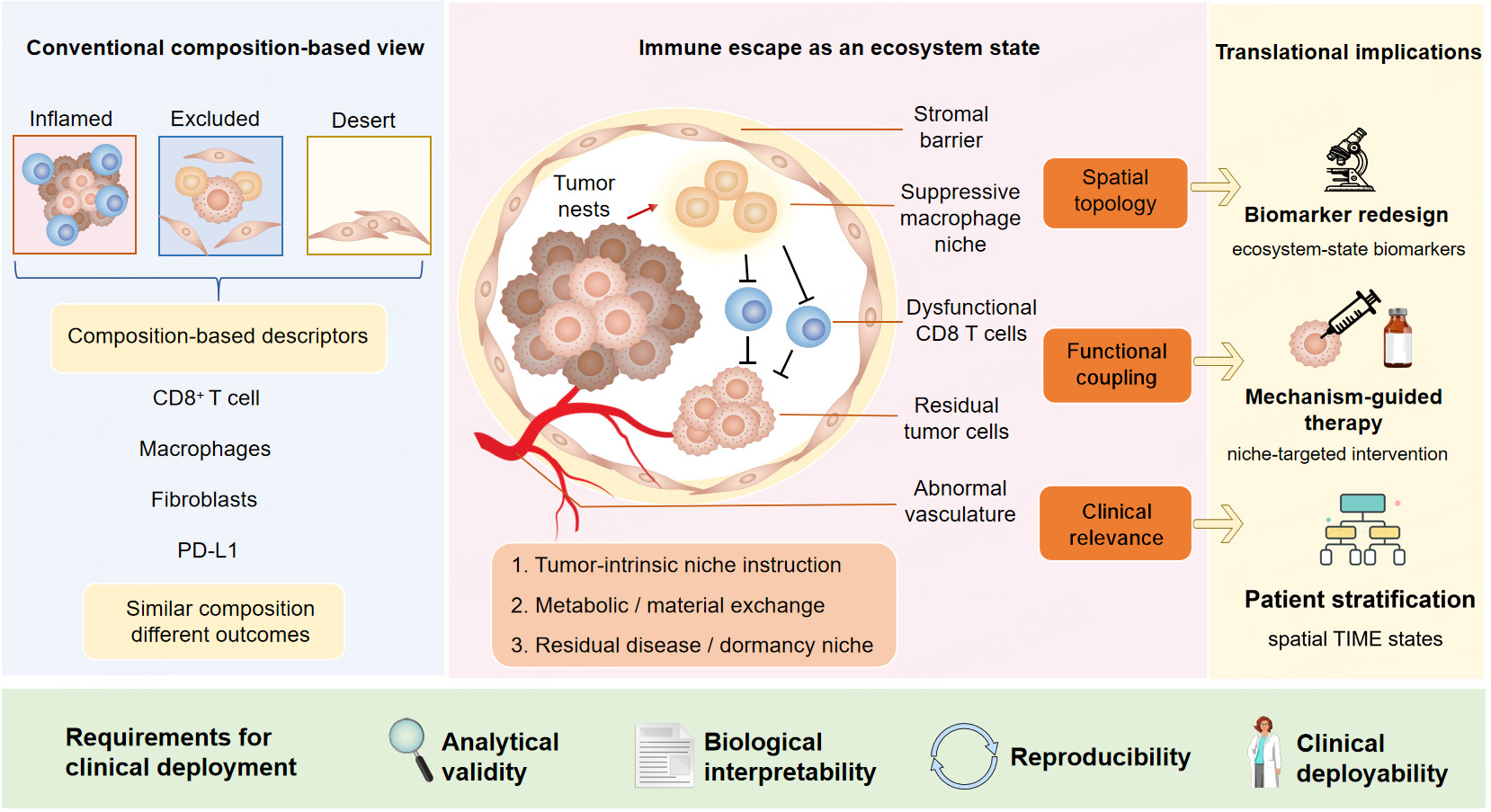
Figure 1. From composition-based TIME descriptors to immune-escape ecosystems. TIME: Tumor immune microenvironment.
Traditional research mainly describes TIME by cell composition and classical phenotypes, including inflamed, excluded, and immune desert tumors. The proposed immune-escape ecosystem emphasizes three core attributes: spatial topology, functional coupling among heterogeneous cells, and definite clinical relevance. In this ecosystem, multiple components, including stromal barriers, tumor nests, suppressive macrophage niches, dysfunctional CD8+ T cells, abnormal vasculature, fibroblasts, and residual tumor cells, interact synergistically.
Similar immune cell composition may lead to totally distinct clinical and functional outcomes, which is driven by three key underlying mechanisms: (1) intrinsic niche regulation derived from tumor cells; (2) metabolic and material exchange within the local microenvironment; (3) formation of niches for residual tumor cells and tumor dormancy.
This new conceptual framework further promotes the redesign of biomarkers, shifting from single indicators (e.g., PD-L1) to ecosystem-state biomarkers. It supports precise patient stratification, mechanism-guided targeted therapy and niche-oriented intervention. For clinical implementation, qualified ecosystem-state biomarkers need to meet four essential requirements: analytical reproducibility, biological validity, clinical interpretability and practical deployability.
For this concept to be valuable, it must signify more than simply “a complicated microenvironment”. An immune-escape ecosystem can be defined by three features. First, it is topological: the pertinent biological processes depend on the patterned spatial arrangement rather than solely on cellular abundance. Second, it is functionally coupled: the constituent populations do not merely coexist but engage, either directly or indirectly, in a coordinated process that undermines antitumor immunity. Third, it holds clinical significance, as the state influences resistance, persistence of residual disease, tumor dormancy, and treatment failure, rather than merely serving as a descriptive snapshot.
What is new in the ecosystem state concept? First, it is not a mere renaming of “spatial heterogeneity” or “immune exclusion”. The novelty lies in requiring simultaneous presence of three criteria (topology, functional coupling, clinical consequence)-most existing descriptions meet only one or two. Second, it offers an explanatory advantage by resolving why composition-matched tumors behave differently: the ecosystem state captures the emergent functional failure that no single marker or cell type can represent. Third, it provides a unifying framework for seemingly disparate mechanisms (p53, L-34, macrophages, mitochondrial transfer, residual disease niches, microbiome topology) as alternative wiring diagrams leading to the same ecosystem outcome.
The proposed “immune-escape ecosystem” differs from existing concepts in several ways. Unlike “immune-excluded” or “inflamed” phenotypes (which are still compositionally anchored), the ecosystem state requires demonstration of functional coupling across distinct cell types. It also differs from “spatial immune niches” (which are often descriptive) by emphasizing clinical consequence as a defining property. Furthermore, in contrast to “ecological tumor states” (which may include proliferation or metabolism), the ecosystem state is specifically defined by immune surveillance failure. Finally, unlike “spatial biomarkers” (which are correlative), the ecosystem state implies perturbability-altering the state changes disease behavior.
This definition clarifies why composition-based thinking now appears inadequate. A tumor exhibiting abundant T-cell infiltration may nevertheless remain poorly controlled if these cells are spatially segregated, metabolically constrained, or situated within immunosuppressive stromal regions. Conversely, a less extensive infiltrate may still hold clinical significance if it resides within an architecture that allows for sustained tumor-immune interaction. Composition is not irrelevant, but becomes interpretable only within its context. This conclusion aligns with recent studies on the TIME and responses to checkpoint blockade, both of which emphasize multicellular organization, rather than solely cell counts, as the level at which clinically relevant biology manifests[2,3].
REPRESENTATIVE EXAMPLES
An effective framework should be capable of organizing representative findings throughout the field. The examples below were selected not for their novelty alone, but because each illustrates a distinct pathway to the same ecosystem outcome: (1) tumor-intrinsic genetic lesion (p53 inactivation) acting through niche construction; (2) metabolic crosstalk (mitochondrial transfer) as a spatially organized suppressive mechanism; (3) residual disease persistence via a targetable macrophage-stem cell niche; (4) patient stratification by immune-evasion programs (colorectal cancer single-cell atlas); (5) ecological topology of an external compartment (gut microbiome). Collectively, they span genetic, metabolic, cellular, and microbial dimensions, supporting the generalizability of the ecosystem framework across cancer types and therapeutic contexts.
One example is the tumor-intrinsic regulation of the surrounding niche. A previous study demonstrated that p53 inactivation in liver cancer induces IL-34 secretion, which subsequently orchestrates the reprogramming of tumor-associated macrophages and facilitates immune evasion by suppressing CD8+ T cell function[7]. The significance of this work extends beyond the pathway itself. The findings indicate that the immunologic effects of a tumor-intrinsic lesion are frequently mediated by local niche construction rather than solely through cell-autonomous immune evasion.
A second example is metabolic dysfunction manifested spatially. A 2025 study published in Nature demonstrated that tumor cells can facilitate immune evasion by transferring mitochondria within the tumor microenvironment, thereby directly impairing immune cell function and reducing the effectiveness of checkpoint inhibition[8]. This advances the concept of metabolic crosstalk beyond the conventional framework of nutrient competition or lactate accumulation. It situates immune suppression within structured local interactions and supports the perspective that immune failure is frequently orchestrated within the tissue environment rather than encoded in a single signaling pathway. Beyond mitochondrial transfer, tumor metabolism impairs CD8+ T cell antitumor activity through multiple spatially organized mechanisms, including lactate acidosis, glucose deprivation, and lipid accumulation, which collectively suppress T cell activation and effector function[9]. Concurrently, chromatin structure and epigenetic regulation influence tumor immunogenicity and immune escape; for example, EZH2-mediated histone methylation can silence antigen presentation machinery and chemokine expression, reducing T cell infiltration and creating stable “epi-niches” that persist even after treatment[10,11].
A third example is derived from minimal residual disease. In hepatocellular carcinoma, a spatial analysis published in Nature Cancer in 2024 demonstrated that PD-L1-positive macrophages sustain residual malignant states through a targetable interaction with stem-like cancer cells[12]. Complementing this spatial analysis, an independent study further demonstrated that Yes-associated protein (YAP) expression status independently predicts post-resection recurrence and poor prognosis in hepatocellular carcinoma (HCC), underscoring the clinical relevance of ecosystem-based stratification[13]. When interpreted in this manner, recurrence cannot be adequately characterized as the survival of intrinsically resistant cells. It is more accurately understood as persistence within a locally organized, immune-permissive niche. The same reasoning applies to dormancy. Recent conceptual and experimental studies have highlighted that dormant disseminated tumor cells evade endogenous immunity not merely due to their quiescence, but because their immune visibility is modified by the local microenvironment[14,15]. In lung adenocarcinoma, stimulator of interferon genes (STING) signaling has been demonstrated to inhibit the reactivation of dormant metastases, highlighting that relapse may represent a change in the ecosystem state rather than simply the removal of an intrinsic proliferative restraint[15].
A fourth example is derived from colorectal cancer. In 2024, an integrative single-cell analysis published in Nature Cancer stratified patients based on distinct immune-evasion programs rather than considering immune escape as a single property of the disease[16]. This study is significant because it demonstrates that ecosystem states can categorize patient heterogeneity into clinically interpretable groups. The microbiome applies the same principle beyond the tumor bed itself. In a 2024 study published in Cell, Derosa and colleagues demonstrated that the ecological topology of the gut microbiota could be translated into a scoring framework associated with immunotherapy outcomes[17]. The conceptual lesson extends beyond the microbiome alone: clinically meaningful biology often reflects ecological structures rather than isolated components.
Collectively, these examples support a practical conclusion. Immune escape is often maintained not by a single lesion or pathway, but by an organized tissue state in which multiple elements mutually reinforce each other. These diverse mechanisms converge on a common ecosystem state, indicating that the framework is disease-agnostic. The concept of the ecosystem does not eliminate mechanistic diversity, it provides a more coherent framework for its interpretation.
WHY CURRENT BIOMARKERS REMAIN INSUFFICIENT
The translational implications are promptly evident. Biomarker development in immuno-oncology continues to depend largely on measures of abundance or average expression, such as PD-L1, mismatch repair deficiency or microsatellite instability, and tumor mutational burden in specific contexts[18,19]. These markers remain clinically significant, but none fully encapsulates the tissue organization that frequently determines whether immunity is effective or locally neutralized. Two tumors may exhibit similar levels of immune infiltration yet respond quite differently, as one maintains accessible effector circuits while the other is structured around suppressive stromal or myeloid components. Similarly, a favorable bulk immune signature may obscure the fact that the critical suppressive program is concentrated in a relatively small yet strategically significant region[5,20].
A more effective framework would aim to identify ecosystem states rather than isolated indicators. This does not imply that all clinical assays must attain maximum complexity or be entirely spatial. The objective should be more practical: to derive tractable readouts that accurately reflect biologically coherent tissue states. An informative biomarker may indicate an immune-excluded fibrovascular architecture, a suppressive macrophage-stromal niche, or a metabolically compromised effector compartment corresponding to a specific therapeutic vulnerability[5,18,19]. Recent pan-cancer and disease-specific studies substantiate this approach by demonstrating that spatial and compositional features enhance response modeling when considered jointly rather than independently[20,21].
Concrete examples of ecosystem-state biomarkers in clinical workflows
To illustrate how an ecosystem-state biomarker might add value to current clinical workflows, consider the following scenarios. First, PD-L1 immunohistochemistry remains a standard companion diagnostic for immune checkpoint blockade, yet its predictive power is limited. A tumor with high PD-L1 expression may still fail to respond if CD8+ T cells are spatially excluded from the tumor nest. An ecosystem biomarker that quantifies the distance between PD-L1+ cells and CD8+ T cells, such as a “spatial exclusion index”, could help identify such non-responders. Conversely, a tumor with low or heterogeneous PD-L1 expression but close physical contact between CD8+ T cells and tumor cells might still derive benefit.
Second, microsatellite instability-high (MSI-H) or mismatch repair-deficient (dMMR) tumors generally respond favorably to checkpoint inhibitors, but a notable fraction exhibit primary resistance. The mechanisms of resistance are often microenvironmental rather than tumor-intrinsic. An ecosystem signature that identifies a suppressive macrophage-fibroblast niche, even within an MSI-H tumor, could potentially stratify patients unlikely to respond to monotherapy, thereby guiding combination strategies.
Third, tumor mutational burden (TMB) is a continuous metric that frequently overlaps between responders and non-responders. Incorporating a spatial neighborhood signature, such as the enrichment of regulatory T cells and PD-L1+ macrophages in proximity to proliferative tumor cells, may improve prediction accuracy. A TMB-high tumor with a spatially “cold” architecture (immune cells largely confined to distant stroma) could behave more like a TMB-low tumor.
Fourth, unlike conventional multiplex immunohistochemistry panels that primarily report cell counts, an ecosystem-state biomarker incorporates predefined spatial rules, such as the proportion of CD8+ cells located within 20 µm of tumor cells or the clustering score of suppressive myeloid cells.
These examples are not intended to discard existing clinically useful assays. Rather, they illustrate how adding spatial and topological information could enhance the predictive accuracy of current biomarkers in real-world workflows. This possibility is increasingly recognized in the spatial oncology literature[3,5].
WHY CLINICAL TRANSLATION REMAINS DIFFICULT
If the ecosystem framework is conceptually compelling, why has it not yet resulted in the development of routine clinical tools? The primary challenges are as much methodological as they are biological. Spatial datasets continue to be susceptible to region-of-interest bias, segmentation errors, platform-specific effects, annotation variability, and algorithm-dependent instability[5,6]. Even when studies arrive at similar conclusions, the analytical approaches employed are often heterogeneous. This limits reproducibility and complicates the distinction between conserved biological signals and method-dependent artifacts.
A second challenge lies in the disparity between observation and intervention. Numerous studies have produced compelling maps of suppressive niches, but visual coherence and association with outcomes are insufficient. Ecosystem features become truly significant when they can be experimentally perturbed and correlated with measurable changes in immune function or therapeutic response. A niche is clinically significant not because it appears convincing, but because its disruption alters disease behavior[7,8,12]. Complicating matters further, therapeutic interventions including immunotherapy, targeted therapy, chemotherapy, and radiotherapy actively remodel the tumor ecosystem rather than simply eliminating tumor cells. Treatment can induce functional reprogramming of tumor-infiltrating immune cells and stromal cells, generate tumor-educated cells with new suppressive activities, and alter metabolic programs in surviving immune cells, all of which may cooperate to drive acquired resistance[22,23]. From an ecosystem perspective, therapy failure often reflects not the intrinsic resistance of a single clone but rather an adaptive shift in the tissue state, representing a remodeling of the ecosystem itself. This observation reinforces the importance of monitoring ecosystem dynamics throughout treatment, rather than solely at baseline, and of considering combination strategies that disrupt the emerging suppressive niche.
A third challenge is translational scale. High-dimensional profiling has broadened the scope of observations, but clinical adoption will depend on simplification. The field is unlikely to benefit from the direct integration of maximal analytic complexity into routine care. A more plausible approach is a staged model wherein high-resolution datasets identify robust ecosystem states, subsequently guiding the development of parsimonious assays, digital pathology readouts, or focused multiplex panels that align with actual pathology workflows[5,6,20,21].
A practical roadmap for ecosystem-based biomarkers
A deployable ecosystem biomarker could take several forms depending on available technology and clinical context. First, a multiplex immunofluorescence panel can be designed with six to eight markers, such as CD8, CD68, PD-L1, α-SMA, E-cadherin, cytokeratin, and 4’,6-diamidino-2-phenylindole (DAPI). Spatial neighborhoods are then defined by predefined rules, for example, quantifying the fraction of CD8+ T cells located more than 50 micrometers away from cytokeratin-positive tumor cells.
Second, a digital pathology score can be developed using deep learning applied to routine hematoxylin and eosin slides. This approach infers spatial exclusion patterns without additional staining, such as a “lymphocyte exclusion index” that has been retrospectively validated in colorectal cancer cohorts.
Third, a spatial neighborhood signature can be derived from spatial transcriptomics data. This composite score captures the relative enrichment of suppressive multicellular hubs, for instance, macrophage-fibroblast aggregates that correlate with poor immune checkpoint blockade response.
Fourth, an integrated clinicopathologic model can combine routinely available immunohistochemical markers, such as PD-L1 and CD8, with straightforward spatial metrics like the tumor-stroma border distance or the proportion of CD8+ cells within the tumor nest.
Each of these candidates requires prospective validation in independent clinical cohorts. Nevertheless, they demonstrate how ecosystem thinking can produce practical and clinically feasible assays without requiring the utmost technological complexity.
PRIORITIES FOR THE NEXT PHASE
The next phase of TIME research should consequently be evaluated less by the complexity of its atlases and more by its capacity to define reproducible, biologically grounded, and clinically applicable ecosystem states. Three priorities are outlined below. The first is standardization: implementing more consistent sampling strategies, annotation frameworks, and endpoint definitions across cohorts and platforms. The second is functional anchoring, which involves a stronger integration of spatial observation with perturbation-based validation. The third is clinical reduction: deliberate efforts to translate complex, high-dimensional ecosystem states into assays that can be implemented outside of specialized research environments[5,6,20].
Recent guidelines, consensus statements, and perspectives concur in their recommendations. The field no longer lacks credible biological insights; rather, it lacks a structured framework for determining which spatial and ecological findings merit translation into clinical tools[18,19,24,25]. A useful ecosystem model should therefore satisfy at least four criteria: analytical validity, biological interpretability, reproducibility across platforms and cohorts, and practical deployability. Recent studies on dormancy and therapy-induced TIME remodeling highlight a common translational bottleneck[26-28]. Studies on intratumoral microbiota and gastrointestinal spatial omics underscore the necessity of advancing from descriptive richness to clinically applicable models[29-33].
POTENTIAL LIMITATIONS OF THE ECOSYSTEM-STATE FRAMEWORK
Despite its conceptual appeal, the ecosystem-state framework has several limitations that warrant acknowledgment.
First, the framework currently relies on post hoc analyses of retrospective datasets; prospective validation in clinical trials is lacking. Whether an ecosystem state can be reliably identified before treatment initiation remains to be established. Second, the required spatial assays, including multiplex imaging, spatial transcriptomics, or high-plex immunohistochemistry, remain expensive, technically demanding, and not widely available in routine pathology laboratories. The disparity between research tools and clinical diagnostics presents a significant obstacle to translation. Third, the framework may be less informative in tumors with uniformly low immune infiltration (e.g., some “immune desert” phenotypes) or in hematologic malignancies where tissue architecture is fundamentally different from solid tumors. Fourth, correlation does not equal causation. Most spatial studies describe associations between ecosystem features and clinical outcomes but do not prove that the ecosystem state drives immune escape. Experimental perturbations, such as niche disruption in preclinical models, are necessary to establish directionality. Finally, an overemphasis on ecosystem complexity may paradoxically impede clinical translation by discouraging efforts to identify simple, parsimonious surrogates. The objective should be to reduce ecosystem states to tractable readouts, rather than to require maximal complexity for every patient.
These limitations do not invalidate the concept but rather highlight priorities for the subsequent phase of research.
CONCLUSION
The most significant advancement in contemporary tumor immunology is not merely the acknowledgment of the complexity of TIME. It is the recognition that immune escape is often most accurately understood as an ecosystem state. Composition matters only within the broader framework of topology, functional coupling, and clinical outcomes.
Once this conceptual shift takes place, several previously disparate lines of evidence begin to converge. Tumor-intrinsic myeloid reprogramming, metabolically mediated immune dysfunction, residual disease niches, dormancy reactivation, and microbiome-linked immune modulation can all be interpreted as distinct pathways leading to a common outcome: a tissue state wherein immune cells are present but no longer capable of exerting effective tumor control[7,8,12,14-17,34].
The field currently demands more than just another comprehensive inventory of immune-evasion mechanisms. A more refined framework is required to determine which tissue states are reproducible, biologically significant, and therapeutically actionable. The future of clinical research on TIME will rely less on cataloging additional components and more on understanding how to identify, measure, and therapeutically disrupt immune-escape ecosystems[1,5,18,19,25].
DECLARATIONS
Acknowledgments
Graphical Abstract created with BioGDP.com[35].
Authors’ contributions
Contributed to the conception of this perspective: Li Z, Yang L
Wrote the manuscript: Yu D, Li Z
Acquired funding: Li Z
Revised the manuscript: Yang L
All authors read and approved the final manuscript.
Availability of data and materials
Not applicable.
AI and AI-assisted tools statement
During the preparation of this manuscript, the AI tool DeepSeek (version R1, released 2025-01-20) was used solely for language editing. The tool did not influence the study design, data collection, analysis, interpretation, or the scientific content of the work. All authors take full responsibility for the accuracy, integrity, and final content of the manuscript.
Financial support and sponsorship
This work was supported by Young Talent of Lifting Engineering for Science and Technology in Shandong, China (No. SDAST2024QTA032), and Wu Jieping Medical Foundation Clinical Research Special Fund (No. 320.6750.2024-17-41).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2026.
REFERENCES
1. Roerden M, Spranger S. Cancer immune evasion, immunoediting and intratumour heterogeneity. Nat Rev Immunol. 2025;25:353-69.
2. Aliazis K, Christofides A, Shah R, et al. The tumor microenvironment’s role in the response to immune checkpoint blockade. Nat Cancer. 2025;6:924-37.
3. Gong D, Arbesfeld-Qiu JM, Perrault E, Bae JW, Hwang WL. Spatial oncology: translating contextual biology to the clinic. Cancer Cell. 2024;42:1653-75.
4. Liu Y, Dai Y, Wang L. Spatial omics at the forefront: emerging technologies, analytical innovations, and clinical applications. Cancer Cell. 2026;44:24-49.
5. Williams HL, Frei AL, Koessler T, et al. The current landscape of spatial biomarkers for prediction of response to immune checkpoint inhibition. NPJ Precis Oncol. 2024;8:178.
6. Jing SY, Wang HQ, Lin P, Yuan J, Tang ZX, Li H. Quantifying and interpreting biologically meaningful spatial signatures within tumor microenvironments. NPJ Precis Oncol. 2025;9:68.
7. Nian Z, Dou Y, Shen Y, et al. Interleukin-34-orchestrated tumor-associated macrophage reprogramming is required for tumor immune escape driven by p53 inactivation. Immunity. 2024;57:2344-2361.e7.
8. Ikeda H, Kawase K, Nishi T, et al. Immune evasion through mitochondrial transfer in the tumour microenvironment. Nature. 2025;638:225-36.
9. Liu H, Yang W, Jiang J. Targeting tumor metabolism to augment CD8+ T cell anti-tumor immunity. J Pharm Anal. 2025;15:101150.
10. Burr ML, Sparbier CE, Chan KL, et al. An evolutionarily conserved function of polycomb silences the MHC class i antigen presentation pathway and enables immune evasion in cancer. Cancer Cell. 2019;36:385-401.e8.
11. Peng D, Kryczek I, Nagarsheth N, et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature. 2015;527:249-53.
12. Lemaitre L, Adeniji N, Suresh A, et al. Spatial analysis reveals targetable macrophage-mediated mechanisms of immune evasion in hepatocellular carcinoma minimal residual disease. Nat Cancer. 2024;5:1534-56.
13. Zhou W, Ye F, Yang G, et al. YAP-based nomogram predicts poor prognosis in patients with hepatocellular carcinoma after curative surgery. J Gastrointest Oncol. 2024;15:1712-22.
14. Adam-Artigues A, Valencia Salazar LE, Aguirre-Ghiso JA. Immune evasion by dormant disseminated cancer cells: a Fermi paradox? Cancer Cell. 2024;42:13-5.
15. Hu J, Sánchez-Rivera FJ, Wang Z, et al. STING inhibits the reactivation of dormant metastasis in lung adenocarcinoma. Nature. 2023;616:806-13.
16. Chu X, Li X, Zhang Y, et al. Integrative single-cell analysis of human colorectal cancer reveals patient stratification with distinct immune evasion mechanisms. Nat Cancer. 2024;5:1409-26.
17. Derosa L, Iebba V, Silva CAC, et al. Custom scoring based on ecological topology of gut microbiota associated with cancer immunotherapy outcome. Cell. 2024;187:3373-3389.e16.
18. Trontzas IP, Syrigos KN. Immune biomarkers for checkpoint blockade in solid tumors: transitioning from tissue to peripheral blood monitoring and future integrated strategies. Cancers. 2025;17:2639.
19. Cottrell TR, Lotze MT, Ali A, et al. Society for Immunotherapy of Cancer (SITC) consensus statement on essential biomarkers for immunotherapy clinical protocols. J Immunother Cancer. 2025;13:e010928.
20. Lindsay JR, Altreuter J, Alessi JV, et al. Pan-cancer spatial characterization of key immune biomarkers in the tumor microenvironment. Cell Rep Med. 2025:102418.
21. Greenwald NF, Nederlof I, Sowers C, et al. Temporal and spatial composition of the tumor microenvironment predicts response to immune checkpoint inhibition in metastatic TNBC. Nat Cancer. 2026;7:435-50.
22. Li D, Shao F, Yu Q, et al. The complex interplay of tumor-infiltrating cells in driving therapeutic resistance pathways. Cell Commun Signal. 2024;22:405.
23. Sun L, Zhao G, Wang S, Li N. Immune cell metabolism in cancer drug resistance: advances in target discovery and clinical translation. Chin J Cancer Res. 2025;37:432-45.
24. Tufail M, Jiang CH, Li N. Immune evasion in cancer: mechanisms and cutting-edge therapeutic approaches. Signal Transduct Target Ther. 2025;10:227.
25. Sullivan RJ, Cillo AR, Ferris RL, et al. SITC vision: opportunities for deeper understanding of mechanisms of anti-tumor activity, toxicity, and resistance to optimize cancer immunotherapy. J Immunother Cancer. 2025;13:e011929.
26. Chen D, Wu R, Liu Z, Wei Y, Kuang D. Therapy-induced remodeling of the tumor immune microenvironment: Mechanistic insights and implications for immunotherapy. Chin Med J. 2026;139:1149-67.
27. Cho J. Understanding tumor dormancy: from experimental models to mechanisms and therapeutic strategies. Biomol Ther. 2025;33:770-84.
28. Pan J, Chen S, Jin L, Chen K, Ye Z, Li B. Immunotherapy-driven remodeling of the tumor immune microenvironment: Spatiotemporal heterogeneity and multidimensional dynamics. Biochim Biophys Acta Rev Cancer. 2026;1881:189544.
29. Zhou Q, Zhou L, Chen X, Chen Q, Hao L. Crosstalk between the intratumoral microbiota and the tumor microenvironment: new frontiers in solid tumor progression and treatment. Cancer Rep. 2024;7:e70063.
30. Liu W, Li Y, Wu P, et al. The intratumoral microbiota: a new horizon in cancer immunology. Front Cell Infect Microbiol. 2024;14:1409464.
31. Zhong L, Li Q, Xiong T, et al. Spatial omics in gastrointestinal oncology: recent advances, therapeutic insights, and clinical translation. J Cancer. 2026;17:515-23.
32. See JE, Barlow S, Arjumand W, DuBose H, Segato Dezem F, Plummer J. Spatial omics: applications and utility in profiling the tumor microenvironment. Cancer Metastasis Rev. 2025;44:87.
33. Lv B, Zhao Y, Li G, et al. Tumor-resident intracellular bacteria scavenger activated in situ vaccines for potent cancer photoimmunotherapy. Adv Healthc Mater. 2025;14:e2404271.
34. Wang B, Ding H, Li Y, Zhao X, Ge P. Immune-excluded and immune-suppressive tumor microenvironments: mechanisms, spatial biomarkers, and therapeutic rewiring. Front Oncol. 2026;16:1855429.
Cite This Article
How to Cite
Download Citation
Export Citation File:
Type of Import
Tips on Downloading Citation
Citation Manager File Format
Type of Import
Direct Import: When the Direct Import option is selected (the default state), a dialogue box will give you the option to Save or Open the downloaded citation data. Choosing Open will either launch your citation manager or give you a choice of applications with which to use the metadata. The Save option saves the file locally for later use.
Indirect Import: When the Indirect Import option is selected, the metadata is displayed and may be copied and pasted as needed.
About This Article
Copyright

Data & Comments
Data
0












Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at [email protected].